
Table of contents
Being well-prepared for FDA inspections is essential for maintaining product quality, regulatory compliance, and avoiding serious business setbacks. FDA inspections are critical events that can shape a company’s market access and reputation.
Redica Systems offers the most comprehensive database of FDA inspection documents, which also powers our Quality and Regulatory Intelligence (QRI) platform. This FDA inspection database provides insights from historical FDA inspections, including Form 483s, 483Rs, Establishment Inspection Reports (EIRs), and Warning Letters. With such comprehensive resources, companies can learn from past experiences, recognize areas of improvement, and better understand the expectations of FDA investigators.
Access the FDA Inspection Database
Understanding an FDA Inspection Database
What is an FDA Inspection Database?
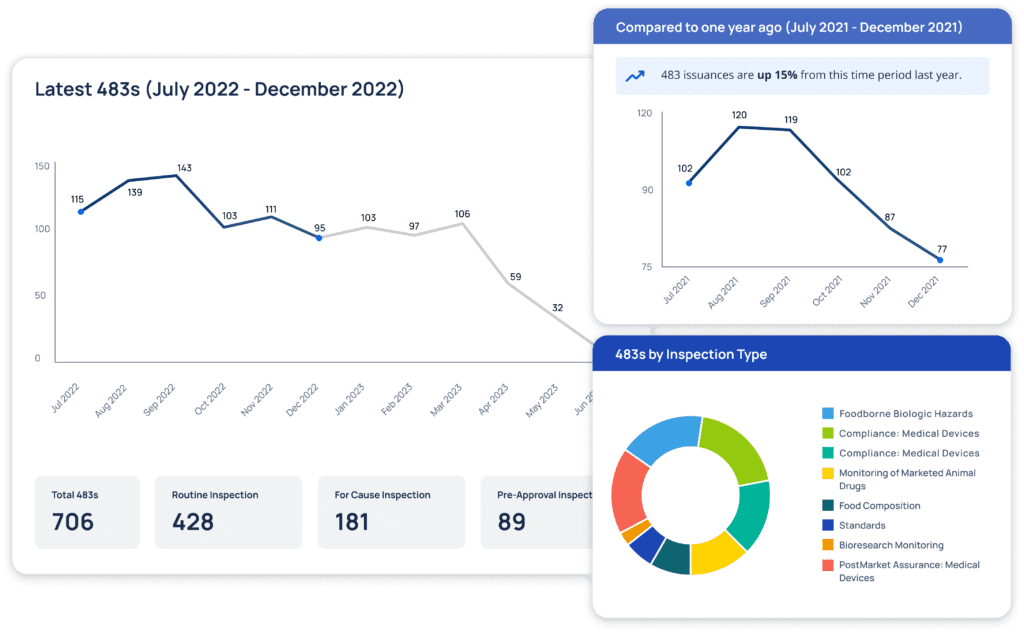
An FDA inspection database is a centralized repository that contains detailed records from FDA inspections. These records typically include critical documents such as Form 483s, 483Rs, EIRs, and Warning Letters. Form 483s are issued by FDA investigators when they observe conditions that may violate the Food, Drug, and Cosmetic (FD&C) Act and other related laws and FDA guidance. EIRs provide a detailed account of the investigator’s findings, while Warning Letters indicate more serious issues that may require immediate action.
The FDA itself offers the FDA Data Dashboard, but it doesn’t make every document available. For more details on the differences between Redica Systems and the FDA Data Dashboard, read How Redica Systems Goes Further than the FDA Data Dashboard.
Overview of FDA Inspection Record Types
Form 483s: These are notices given to companies when FDA investigators observe any potential regulatory violations. They provide an opportunity for companies to rectify issues before further regulatory action is taken.
483 Responses (483Rs): The company receiving the 483 can respond to FDA. This response in our database is called a 483R. These are valuable for research and preparation purposes because they show how the recipient of the 483 plans to address the issues highlighted.
Establishment Inspection Reports (EIRs): These documents are official reports that summarize the results of an inspection, including observations, discussions with management, and responses.
Warning Letters: Issued when significant regulatory non-compliance is identified, Warning Letters are a serious call to action for manufacturers to address deficiencies. Warning Letters often follow a bad 483 if FDA is not confident that the problems will be promptly addressed and are not likely to reoccur.
In addition to the four main inspection record types above, an FDA inspection database should provide Final Inspection Classification which summarizes the overall outcome of an inspection, classified as No Action Indicated (NAI), Voluntary Action Indicated (VAI), or Official Action Indicated (OAI). A VAI classification indicates FDA is confident that the company can address the deficiencies detailed in the 483, while an OAI status indicates more formal action is needed — often indicating a Warning Letter is forthcoming.
Utilizing an FDA inspection database, as part of a broader Quality and Regulatory Intelligence capability, provides life sciences companies visibility into past inspection outcomes and helps identification of trends that may apply to their own operations. It helps better prepare for their own future inspections, as well as make more informed decisions about supply chain partners. They are key tools in reducing compliance risk and improving product quality.
Recommended Reading: FDA 483 Availability and How to Get Them
Preparing for FDA Inspections
FDA inspections can be challenging and thorough preparation is key to success. Understanding the different types of FDA inspections and leveraging available guidelines will help companies ensure compliance.
Overview: The 4 Types of FDA Inspection
FDA inspections can vary based on the product, facility, and regulatory requirements. There are four main types of FDA inspections:
- Pre-Approval Inspections (PAI): Conducted to verify the accuracy of data submitted in drug applications and to evaluate manufacturing facilities.
- Surveillance Inspections: Routine inspections conducted to ensure ongoing compliance.
- For-Cause Inspections: Targeted inspections following reports of adverse events or manufacturing issues. There are also “Post-Approval Inspections” sometimes called “Follow-Up Inspections.”
- Bioresearch Monitoring (BIMO) Inspections: Conducted to evaluate data integrity, clinical research practices, and compliance with regulations.
Understanding the purpose and scope of each type of inspection allows companies to prepare effectively.
Recommended Reading: Understanding FDA Inspection Guidelines
Inspection Classification and Risk Assessment
FDA Inspection Classification Overview
Following an inspection, the FDA classifies the outcome into one of three categories:
No Action Indicated (NAI): No significant regulatory violations were found during the inspection.
Voluntary Action Indicated (VAI): Some issues were observed, but they are not significant enough to warrant immediate regulatory action. The company is expected to address these issues voluntarily.
Official Action Indicated (OAI): Significant regulatory violations were observed, and the FDA may take enforcement actions unless corrective measures are taken.
Final Inspection Classification and Its Importance for Compliance
Understanding the final classification of inspections is essential for risk assessment and ensuring ongoing compliance. When inspections result in an OAI classification, companies must act swiftly to implement corrective actions and address issues effectively to avoid further regulatory scrutiny or penalties.
Inspection Classification Database
Leveraging the inspection classification database enables QA and Operations teams to understand broader trends in inspection outcomes. It can also help companies anticipate what type of observations may lead to an “Official Action Indicated” outcome and prepare accordingly. By assessing patterns, pharma, and medtech companies can implement preventive measures to mitigate the risk of receiving an OAI classification.
Utilizing tools such as our Quality and Regulatory Intelligence Platform and our FDA Inspection Database will provide the insights and data necessary to achieve these goals.
Frequently Asked Questions About FDA Inspections
Are FDA Inspection Reports Public?
Yes, FDA inspection reports such as Establishment Inspection Reports (EIRs) can be requested through the Freedom of Information Act (FOIA). However, the process may take time, and certain proprietary information may be redacted. The FDA makes certain documents readily available on https://datadashboard.fda.gov/ora/index.htm.
Are FDA 483s Publicly Available?
Yes, as above.
When can a firm expect to receive the final inspection classification from the FDA?
The time it takes the agency to arrive at a final classification and communicate it depends on many factors, including evaluation of information collected during an inspection, as well as information provided by the facility following an inspection. Inspection classification recommendations from investigators are an important factor in the evaluation and in the classification process. Additional information is considered to determine the final inspection classification, including the facility’s response to inspectional observations highlighted in Form FDA 483 and the company’s proposed or completed corrective actions.
What types of inspections are not included in the database?
Not all inspections are included in the database. Those not included are:
- Inspections conducted by states, pre-approval inspections, mammography facility inspections, inspections waiting for a final enforcement action, and inspections of nonclinical labs are not included.
- Inspections of nonclinical labs are available at non-clinical laboratories inspected under good laboratory practices.
What are the benefits for consumers and firms in sharing the FDA’s inspection information?
Sharing redacted inspection information helps contribute to company transparency and educates consumers about what the company is doing to address FDA concerns.
What can a regulated small business do if action is indicated after an inspection?
Pay strict attention to what FDA found and is requesting the company to do and consider hiring industry consultants to help with remediation efforts and communication with the agency.
