
Table of contents
Updated December 2024
Introduction to the MAUDE Database
The FDA regulates a wide spectrum of healthcare products, ensuring that medical devices, pharmaceuticals, and other health-related goods entering the market meet stringent standards of quality, safety, and efficacy. Central to this mission is the FDA’s Manufacturer and User Facility Device Experience Database or MAUDE.
What is the MAUDE Database?
The MAUDE database is a publicly accessible platform maintained by the FDA that compiles Medical Device Reports (MDRs) submitted under 21 CFR 803. These reports detail adverse events where a device may have caused or contributed to death, serious injury, or malfunction. By offering visibility into postmarket performance and patient safety outcomes, the MAUDE database serves as an essential resource for industry decision makers. The FDA deserves considerable recognition for its role as perhaps the most transparent and informative health agency worldwide, making valuable data, including the MAUDE database, accessible to the public despite the significant effort and cost involved.
For decision makers in the pharmaceutical and medical device industries, effectively utilizing and interpreting MAUDE data is key. This pillar page explores best practices, strategies for successful FDA site inspections, and methods for leveraging the MAUDE database to inform compliance, design enhancements, and postmarket surveillance efforts.
MAUDE Database: Purpose, Structure, and Scope
MAUDE Reporting Requirements
Under the Medical Device Reporting regulation (21 CFR 803), specific entities must submit reports of adverse events, serious injuries, and device malfunctions:
- Manufacturers: Required to report instances where their devices may have caused or contributed to death or serious injury. They must also report any malfunction likely to recur and result in serious outcomes.
- Importers: Required to notify both the FDA and the manufacturer for device-related deaths or serious injuries, and to alert manufacturers regarding device malfunctions with potential serious outcomes.
- Device User Facilities (e.g., hospitals, outpatient facilities): Must report suspected device-related deaths to both the FDA and the manufacturer. Serious injuries must be reported to the manufacturer and, if unknown, to the FDA.[1]
These submissions, along with voluntary reports from patients, healthcare professionals, and other public stakeholders, populate the MAUDE database. This broad range of sources ensures that the MAUDE data reflects a diverse set of clinical experiences and device usage scenarios.
Limitations and Considerations When Using MAUDE Data
While the MAUDE database offers a substantial pool of information, it is essential to understand that the data is not independently verified by the FDA. Reports may be incomplete or contain inaccuracies, and the database does not provide information on the frequency or volume of device use[2]. In addition, although reports should be filed within specified timelines (5, 10, or 30 days in accordance with § 803.53(b)), delays are common and unavoidable when initial data is incomplete or not properly reported to the manufacturer. Decision makers should interpret MAUDE data with a critical eye, supplementing these reports with internal complaint data, market intelligence, and academic literature.
Leveraging the MAUDE Database for Risk Management and Quality Improvement
Filtering and Benchmarking Strategies
Filtering the MAUDE data by product family, device type, manufacturer, or date range helps reduce information overload. Benchmarking device performance against competitor products or industry norms also provides valuable context. Leveraging advanced analytics and visualization tools can streamline data interpretation, enabling decision makers to focus on insights that drive meaningful improvements rather than sifting through irrelevant or redundant reports.
Quantitative Analysis of MAUDE Data
Quantitative analysis involves identifying patterns, trends, and frequencies of reported adverse events over time. By systematically reviewing the number and type of events associated with a particular device, technology, or product family, compliance teams can detect sudden increases in reporting that may signal product quality issues or emerging risks. Metrics to consider include:
- Event Frequency and Rate Over Time: Monitoring changes in the rate of adverse events helps organizations identify potential device-related problems before they escalate.
- Geographic or Demographic Trends: Identifying whether certain regions or patient populations are associated with higher reporting volumes enables targeted investigations and mitigation efforts.
Qualitative Analysis of MAUDE Data
Qualitative review complements quantitative insights by examining the content and context of individual reports. Through careful reading of event narratives, companies can gain a deeper understanding of the circumstances surrounding device malfunctions or injuries. This may involve:
- Identifying Recurrent Failure Modes: Highlighting common device components or processes that may be prone to issues.
- Understanding Patient and Environmental Factors: Assessing how patient comorbidities, clinical settings, or usage conditions might influence outcomes.
- Providing Input for Design Controls: Informing product design modifications, packaging improvements, or labeling enhancements to mitigate identified risks.
Ensuring Compliance with Postmarket Surveillance Obligations
Global Compliance Perspectives
Global Compliance teams leverage MAUDE data to benchmark an organization’s devices against competitor products and broader industry trends. By continuously monitoring and analyzing events, quality teams can prepare for FDA inspections, anticipate questions, and confirm that existing quality management systems align with regulatory expectations. In Redica’s linked data models, inspections linked to postmarket events are used to ensure continuous inspection readiness.
Postmarket Reporting Integration
In an effective postmarket surveillance program, MAUDE data is not evaluated in isolation. Instead, it is integrated into a broader set of reporting mechanisms governed by quality system procedures (e.g., ISO 13485, ISO/TR 20416) and regulatory guidelines. By synthesizing external MAUDE reports with internal complaint logs, product-specific risk assessments, and reliability data, organizations can develop a clearer, more holistic view of device performance. In the Redica database, specialized filtering, search, and visualizations are provided to ensure that appropriate product and procedural benchmarking can occur.
Risk Management Applications
MAUDE data can inform risk management activities from the early phases of device development through postmarket surveillance. By correlating known hazards and harms with observed adverse events, companies can refine their risk profiles, prioritize corrective and preventive actions, and implement targeted design controls. Using aggregated MAUDE insights enables a balanced, evidence-based approach to product safety and reliability, reinforcing the foundation for successful FDA inspections.
Redica Systems provides data visualization tools such as charts, graphs, and dashboards, to monitor and summarize the large volume of data contained in MAUDE and highlight trends or outliers. We utilize methods such as time-series analysis and correlation analysis to identify relationships between different variables and assess the significance of observed patterns. Real-time data pipelines ensure that data is available upon publication and continuous monitoring can occur.
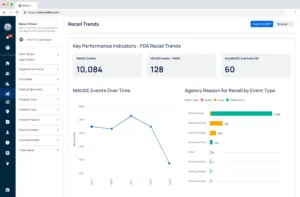
Figure 1 | Screenshot of Redica Systems – Key Performance Indicators – FDA Recall Trends
Challenges and Solutions
When interpreting MAUDE data, it’s crucial to consider the context and limitations of the information provided as well as the queries being made of the data. As mentioned earlier, MAUDE reports are not verified by FDA and may contain incomplete or inaccurate information; context is key for interpreting the results. Additionally, the number of reports for a particular device does not necessarily reflect the actual incidence of adverse events, as reporting rates can be influenced by factors such as device usage, market share, and reporting practices.
With regard to querying the data correctly, ensure the right filters are being used to generate useful outputs. For example, a request for site-level data can give you insights about historical and current reporting volumes and may be indicative of quality issues, but it won’t give you information about a particular product or event. Similarly, MAUDE reports cannot in many circumstances be linked to other events such as recalls or inspections until a proper qualitative analysis is performed. These issues in particular can be difficult to explain to industry outsiders, and should always be explained in context with deep understanding of the reports and possible contributing events.
It’s essential to use tools that allow for identification and visualization of the “correct” (i.e. meaningful and useful) data. Then we must supplement that data with other sources of information, such as internal complaint data, scientific literature, and expert opinions, to gain the most comprehensive understanding of the issues at hand.
Conclusion
MAUDE data is a valuable resource for medical device professionals seeking to enhance product safety, quality, and compliance. By understanding the reporting requirements, analyzing the data effectively, and applying best practices, we can leverage this information to drive meaningful improvements in patient care and device performance.
It is crucial to approach MAUDE data with a critical eye and a deep understanding of its regulatory framework. With the appropriate use of MAUDE data in conjunction with other sources of information we can gain deep new insights into product quality and safety. By proactively monitoring adverse events, identifying trends and patterns, and investigating potential issues, we can take swift action to mitigate risks, improve device design, and enhance patient outcomes. In addition, with a commitment to data-driven quality improvement and regulatory compliance, we can build trust with patients, healthcare providers, and regulatory authorities.
[1] This post does not discuss the Voluntary Malfunction Summary Reporting (VMSR) program which allows manufacturers to report certain malfunctions on a quarterly basis as summaries.
[2] A note to industry outsiders: MAUDE data is often misunderstood by those outside of the medical device industry, and we should be clear that a MAUDE report is not necessarily evidence that a device caused an adverse event. Incomplete and conflicting information is reported to manufacturers from many different sources and companies aim to report this information as best they can pursuant to the regulations. Each medical device manufacturer and facility has procedures specifying criteria for reporting within the definitions provided by FDA. These procedures are routinely inspected by FDA to ensure that companies remain compliant with the regulation. Most companies report events “conservatively” meaning that over-reporting of events is common, particularly in products that are used in critically ill or elderly patients. These patients, regardless of treatment or device, often experience many adverse events due to comorbidities which are reported regardless of device causation or contribution.

Alison leads Redica’s data strategy efforts to shape how data quality, structure, and analytics support customers’ compliance and risk detection workflows. Her perspective guides how Redica’s intelligence can be used to solve real-world regulatory and quality challenges at scale.
