.webp)

Table of contents
Share

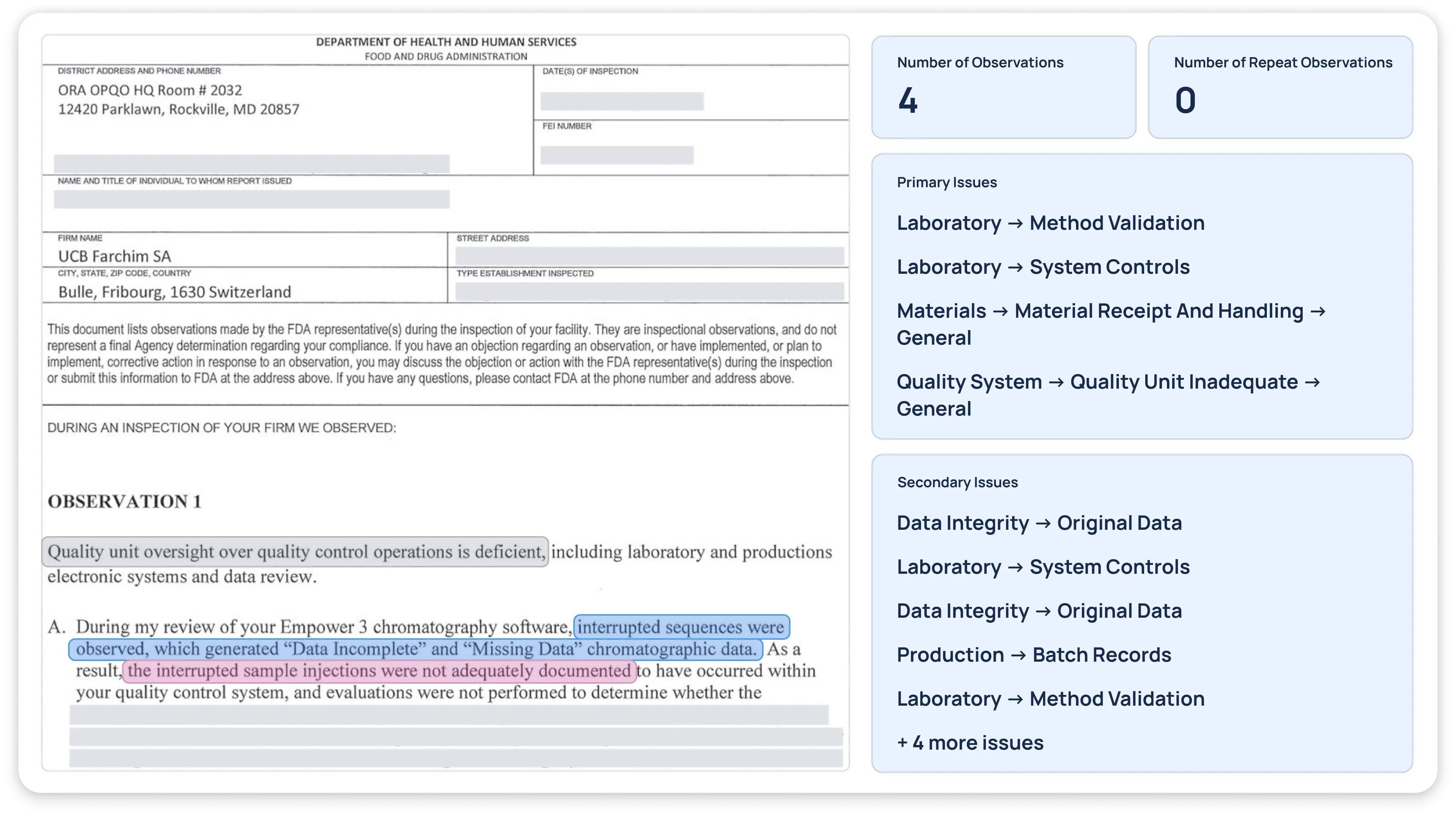
An FDA 483 typically includes the following essential features:Issuing FDA Field Office: Identifies the local FDA district responsible for the inspection.Inspection Dates: Shows the timeline of the inspection, which can last from a single day to multiple weeks.FEI Number: The Firm Establishment Identifier assigned by the FDA’s district office for the inspected facility.Manufacturer’s Contact Information: Details for the company and facility under inspection.List of Observations: The critical portion of the document. Observations can range from just a few to dozens, describing each alleged deviation from regulatory requirements.Investigator Details: The name and title of the FDA investigator(s). Multiple investigators or nationally recognized experts may indicate a significant or complex inspection scope.Interpreting a 483 requires attention to both the scope and the details of each observation:Length and Level of DetailA 483 spanning many pages indicates multiple quality system failures and warrants immediate and comprehensive remediation.Length alone does not always determine severity, but multiple examples of repeated problems or extensive detail often signals serious concerns.Repeat ObservationsWhen an observation references a previous inspection’s findings, it suggests that corrective actions from a prior 483 response either were not implemented effectively or were wholly disregarded.Repeat issues significantly increase the risk of escalation to a warning letter or additional enforcement action.Extent of Supporting ExamplesThe FDA often lists several specific examples to demonstrate a systemic problem.More than three examples under a single observation generally suggests widespread issues rather than isolated defects.Scope of the ObservationsObservations involving data integrity, aseptic practices, or cross-contamination pose direct threats to product quality and patient safety.These observations frequently drive the FDA to seek swift corrective measures or escalate to a warning letter.Investigator ExpertiseIf a nationally recognized expert leads or participates in the inspection, the observations may reflect FDA-wide priorities rather than a single district’s focus.To evaluate the seriousness of a 483 and the potential for additional enforcement action, ask the following questions:Is the 483 longer than 8-10 pages? Longer is not better. That doesn’t mean that a short 483 may not lead to serious enforcement actions, but a long 483 suggests problems may exist in many quality systems. The FDA also concludes that if one quality system is out of control, they are all out of control.Do any of the observations have an extensive description of examples? At the end of 2015, a 483 issued to a firm in China identified 6 ½ pages of examples of how the firm manipulated electronic records. This is a situation where the FDA may be attempting to drive home a point to the firm and to others who read the 483. As a rule of thumb, consider any observation supported by more than 3 examples is one where the agency is trying to send a message about the breadth of the problem within the organization, and may be an indication that additional enforcement actions are in store.Do any of the observations state that they are repeat observations from previous inspections? This statement indicates that while the firm’s response to the previous inspection may have been acceptable, they did not implement effective/corrective/preventive actions to ensure a permanent ‘fix’ for the problem. This type of statement suggests that additional enforcement actions may be forthcoming.Is the 483 issued to an executive? Is the 483 addressed to the CEO of the company rather than the head of the site at which the inspection was conducted? If so, it is yet another suggestion that the FDA is sending a message to the corporation regarding the seriousness of the inspection observations.Do the areas addressed include lack of data integrity and poor aseptic practices with potential for contamination or cross-contamination? This combination of problems, particularly in the past few years, has frequently resulted in a warning letter or additional enforcement action.How many investigators performed the inspection and who were they? If any are national experts, that is important because observations in their areas of recognized expertise often reflect an agency-wide focus and interpretation of requirements.Understanding these attributes offers clues to the gravity of the FDA’s findings. For instance, a 483 addressed directly to a Chief Executive Officer (CEO) instead of a site manager can signal that the FDA is issuing a high-level warning about system-wide compliance issues.Redica Systems’ DocIQ™ feature makes interpreting regulator enforcement documents like 483s easier and faster. Read “Avoiding the Trap of a Data Swamp” to learn more on DocIQ™. Figure 3 | An abstract of Redica Systems’ DocIQ™
about the author(s)
