Table of contents
Cell and gene therapy (CGT) is an emerging and promising field of medicine that offers cures for rare and debilitating diseases, though often at exorbitant costs. The science supporting CGT is complex, and serious side effects are possible.
The complexity and variability of these technologies, their novelty, the assays required, and the excitement of the clinical results that sometimes occur make this emerging field of medicine challenging to regulate.
To that end, FDA recently stood up a new Super Office. The Office of Tissues and Advanced Therapies (OTAT) has now become the Office of Therapeutic Products (OTP) within the Center for Biologics Evaluation and Research (CBER). The agency believes this new office is necessary to meet its growing workload and new commitments under the Prescription Drug User Fee Act (PDUFA VII) agreement for FY2023-2027.
According to the FDA’s website, “the mission of OTP is to promote public health through a data-driven process to provide regulatory oversight that helps ensure medical products are safe and effective. OTP oversees the development and regulation of a wide variety of biological products, including cell therapy products, tissue-engineered products, gene therapy products, plasma protein products derived from blood and their recombinant analogues, and certain medical devices used in the production of these products.”
“In addition to performing regulatory review of product quality, safety, and effectiveness, the Super Office conducts applied scientific research related to the products that it regulates, develops relevant regulatory policies, and supports other agencies and center components involved in ensuring compliance with CBER biologics regulations.”
The recent announcement regarding the new OTP states that “utilizing key tenets of CBER’s modernization efforts, CBER will…elevate OTP to a Super Office to manage its program at a macro level and to better position the Center to address an everchanging public health landscape.”
At the ISPE Annual Meeting held in Orlando, Florida, in early November 2022, just as the change from OTAT to OTP was taking place, OTAT Division of Cellular and Gene Therapies Branch Chief Melanie Eacho discussed her office and the work it does, which will continue under a different name at OTAT. The Cell Therapies Branch focuses on the review of chemistry, manufacturing, and controls information in regulatory submissions for cell therapy products.
“Our office currently is OTAT, and when it becomes OTP, will continue to review a diverse number of cell and gene therapy products,” she explained. Eacho provided the following slide highlighting the diversity of products her office regulates (Figure 1.).
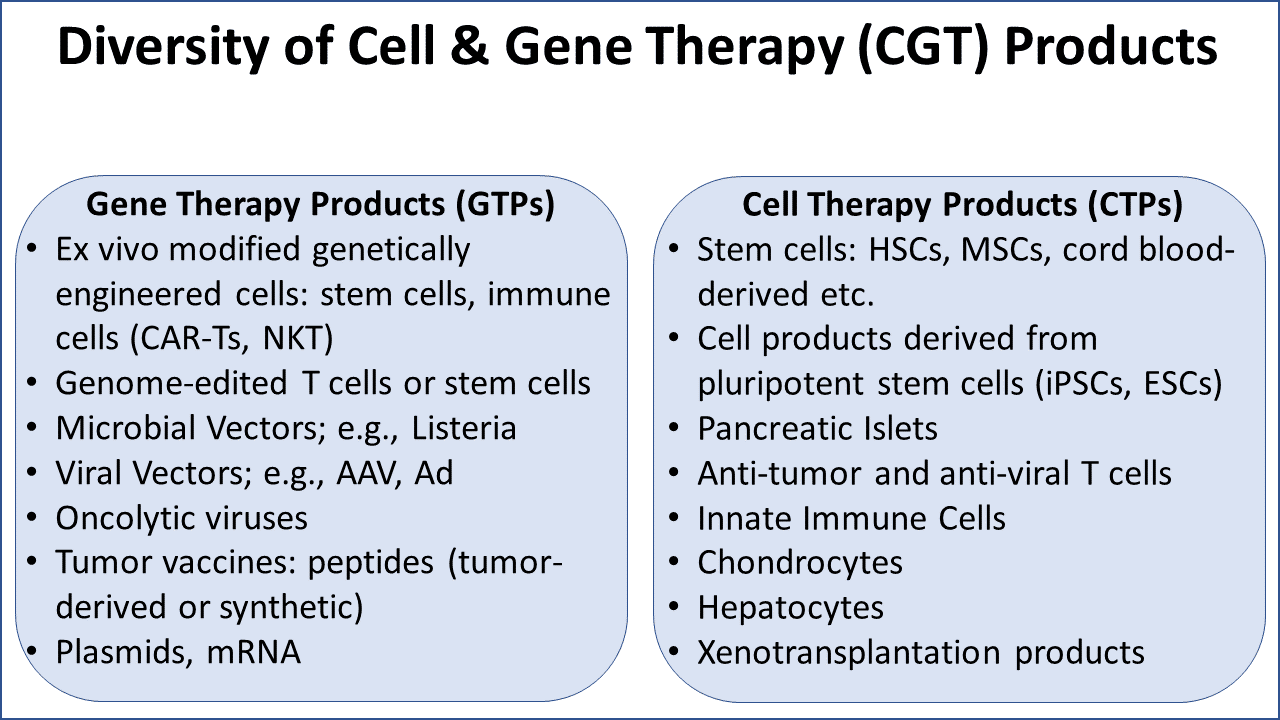
Cell and gene therapy products encompass not only a wide variety of products but also present common concerns, whether it be autologous or allogenic, regarding the complexity of the product and its mechanism of action and all aspects of manufacturing controls.
In addition, there are specific concerns related to the origin of source materials, with the autologous category leading to its own set of specific concerns. (See Figure 2.).

There is a lot of manufacturing information to cover in an original Investigational New Drug (IND) application and to maintain over the span of product development, from early pilot phases to the later pivotal phases of the clinical study. And as development progresses, the initial CMC information gets refined, processed, and locked down to support clinical safety and efficacy data being collected to support marketing licensure.
The amount of information to be submitted will depend on the phase and scope of the initial clinical investigation, which includes a description of the composition, manufacture, and control of the investigational product, and should be sufficient to assure the identity, quality, purity, and potency – biological activity – of the investigational product. Early-phase information should focus more on safety, including identity, purity, and activity, and later phases should focus more on quality and potency.
Stay tuned for three subsequent parts of this article detailing the insights Eacho provided:
- Part Two: Common CMC Issues In Early Development
- Part Three: GCT Product CQAs, Lot Release, Device Considerations
- Part Four: CMC Expectations And Challenges In Late Phase Development.