Table of contents
Common questions across the pharma industry since the business model shifted years ago to contracting out manufacturing and supply chain operations include, “Who is responsible when current Good Manufacturing Practice (CGMP) regulations are not followed? Which entity is subject to enforcement actions by regulatory agencies including 483s and warning letters?”
Those questions were explored by senior FDA officials as part of two podium presentations and a Q&A session at the PDA/FDA Joint Regulatory Conference 2024 held in Washington, DC, September 9-11 2024.
In his presentation on “CDER Updates,” FDA Center for Drug Evaluation and Research (CDER) Office of Manufacturing Quality (OMQ) Office of Compliance (OC) Director Francis Godwin addressed the question, “Does CGMP apply to other entities in the supply chain besides manufacturers?”
He referenced the Preamble to the 1978 GMPs which states in part that “a drug is deemed to be adulterated unless the methods used in its manufacture, processing, packing, and holding, and the facilities and controls used therefor, conform to current good manufacturing practice.”
Another section states that the adulteration provisions apply to “wholesalers, retailers, pharmacies, and hospitals as well as manufacturers.” Godwin presented a series of slides showing which entities are covered, summarized here:
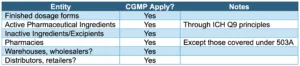
Regarding non-manufacturing supply chain participants, he explained that once a drug or device is in the distribution chain – for example being transported or stored in a warehouse – it can be exposed to conditions that would render it adulterated.
By way of example, the CDER director pointed to a May 2023 FDA notice, “Family Dollar is Initiating a Voluntary Recall of Certain Over-the-Counter Drug Products Because the Products Have Been Stored Outside of Labeled Temperature Requirements.”
“Think of it like this,” he said, “there is chocolate that you get in the mail you order online. If it is going to be sitting in a warehouse in say, Mississippi or Alabama in the height of summer, it is not going to look like what you thought it should when it shows up. That same principle will apply to drugs in the supply chain and CGMP can apply in these instances.”
While FDA does not conduct routine surveillance inspections of non-manufacturing entities, they are subject to for-cause inspections should FDA have reason to believe there may be issues that could impact product quality.
Who Has the Ultimate Responsibility for Product Quality?
Godwin – whose signature appears on CDER warning letters – explained that CGMP contractors are extensions of their clients.
“If you are contracting out a CGMP activity, you need to make sure that that contractor is meeting the applicable CGMPs for what they are doing. This is language that we use in quite a bit of our warning letters: “FDA is aware that drug manufacturers use independent contractors. FDA regards contractors as extensions of the manufacturer, and you are responsible for the quality of the drugs regardless of any agreements you have in place.”
Does FDA Also Hold Contract Facilities Accountable?
In her presentation on “CDER Compliance Updates,” CDER Office of Compliance Director Jill Furman addressed the question around contract facilities.
“When we encounter situations where there are contract manufacturers and multiple players involved, sometimes questions arise regarding who is responsible for drug quality. Is it the contract manufacturing organization performing manufacturing operations on behalf of the manufacturer? Or is it the distributor? Or is it the labeler? Or is it the FDA?”
“The FDA’s role, of course, is to provide proper oversight of regulated industry, but it is ultimately the responsibility of those in the supply chain to comply with the law. Each party engaged in the manufacturing of a drug is responsible for ensuring compliance with CGMP in the manufacturing activities that it performs.”
“We are looking at the entire supply chain,” Furman stressed. “We have also held distributors responsible as appropriate when they distribute adulterated products in the United States.”
She noted that FDA warning letters have cited distributors for failing to have adequate procedures to ensure products met quality attributes and for failing to have adequate supplier qualification procedures.
“The takeaway message here is, wherever you may be in the supply chain, it is important to understand your relationships and who you are working with. I would encourage everyone to read closely the FDA guidance on contracting manufacturing arrangements for drugs.”
Furman provided examples from recent warning letters in which various supply chain participants have been held responsible, and provided her comments.
FDA recently issued a warning letter to a contractor of a single injection eye treatment in which it noted, “you are responsible for the quality of drugs you produce as a contract facility, regardless of agreements in place with application sponsors and product owners” (see Figure 1, Warning Letters to CMO and Domestic Distributor).

Figure 1 | Warning Letters to CMO and Domestic Distributor
“Next are excerpts from warning letters sent to distributors of ophthalmic drugs in 2024. They are notable for the inclusion of two prohibited act charges related to the receipt and delivery in interstate commerce and introduction into interstate commerce of adulterated drugs.”
“In the case of the first distributor here, that one failed to have adequate procedures to ensure that its ophthalmic drug products produced for the firm met appropriate quality attributes and also failed to have that adequate supplier qualification procedures” (see Figure 2, Warning Letters to Ophthalmic Drug Distributors 2024).

Figure 2 | Warning Letters to Ophthalmic Drug Distributors 2024
“One other point to note is we saw a delayed recall action that we cited in a couple of warning letters. This one is an excerpt from a warning letter sent to a distributor and manufacturer of ophthalmic drugs in July, specifically noting that distributor’s delay recalling adulterated drug product” (see Figure 3, Distributor’s Role, Delayed Recall).

Figure 3 | Distributor’s Role, Delayed Recall
Sponsor/Contractor Role Likened to a Marriage
In a Q&A after the session titled, “Current GMP Compliance Trends and Topics,” CBER Office of the Center Director Senior Science Advisor Ingrid Markovic, who served as the moderator, introduced the following question to the panel.
“I heard a lot of talks in the last couple of days about CMOs and referencing the importance of quality agreements. And I think one thing that also came up is that sometimes that relationship could be a little bit strained because there is not really a bright line over who owns the know-how, and what is proprietary and what is not. Do you have any advice for sponsors how to select their CMOs, their contract labs, and how to navigate that contract space?”
CDER’s Francis Godwin replied using an analogy that the relationship between a sponsor and a contractor is like a marriage – an analogy that the other panelists built on.
He commented, “If you are a sponsor, the contract manufacturing guidance talks about product owners who are outside of the application realm. When you select a contract manufacturer, you are marrying them in terms of what FDA sees. And although a quality agreement is not legally required, it is highly recommended to delineate who is in charge of what.”
He explained that “one of the reasons that guidance came out was the agency was seeing a lot of sponsors, product owners, whatever you want to call them, and contractors where they hadn’t delineated who was responsible for what or who conducts inspections and neither party was doing something that needed to be done.”
When FDA sees gaps of that sort, Godwin stressed, “we are not yelling at the sponsor, we are not yelling at the manufacturer, we are yelling at both. We have actually had multiple meetings where we call in the sponsor and the contract manufacturer and say, ‘This relationship is toxic. You are not doing this. You need to fix this.’”
In selecting a CMO, he pointed out, financial implications, purchasing, supply, and other considerations come into play. “But on the quality side, you need to make sure that you are comfortable, that your quality units can actually interact and function cohesively. So don’t let the finance guys try and say, ‘Oh, this is the cheapest manufacturer. It is the lowest bid. Let’s go with them.’ Because then you might end up paying the price later if there are quality problems.”
Other Panelists Weigh in
“I love the marriage analogy there, I will definitely echo that,” said CBER Biological Drug and Device Compliance Quality Branch Chief and Consumer Safety Officer Daniel DeCiero
“I was thinking about it the same way, where apart from ‘for better or for worse,’ there will be issues that come up throughout that relationship. And it is important to have someone that you can work with. I mentioned some issues and challenges with particulates and syringe quality or issues [in my earlier presentation]. Oftentimes, when we are doing an investigation, we want to work with that vendor and supplier to ask, ‘Is this a problem that’s coming from there? Or is this a problem that’s coming from the manufacturing side?’”
“Being able to work with them in a good working relationship, which can include quality agreements and other things of that nature, is absolutely essential to driving quality throughout the entire product lifecycle.”
FDA Office of Pharmaceutical Quality Operations Compliance Branch Director Ronda Loyd-Jones continued the analogy by likening quality agreements to prenuptial agreements.
“Outline what you have with each, what the manufacturer and what the contractor are bringing into this relationship, what their responsibilities are, and what you are getting out of it,” she recommended.
FDA Center for Veterinary Medicines (CVM) Division of Drug Compliance Consumer Safety Officer (CSO) Marea Banks commented on how quality agreements come into play during inspections and when the agency works with firms to help them come into compliance.
For example, she noted cases where CVM reviewed Establishment Inspection Reports (EIRs) in which the investigator explained the agreements between the manufacturer and contract facilities.
“I must say that it is an easier process for the investigators as well as for CVM compliance when we review cases when those types of contracts or quality agreements are laid out in advance with specific criteria. When there is an issue, it is more easily addressed and more efficiently addressed during the inspection.”
It can also help FDA evaluate whether some sort of regulatory action may be needed and whether a firm is capable of achieving some degree of voluntary compliance. “The quality agreement tool is instrumental for the work that I do in compliance,” Banks explained.
She concluded with a reference to the marriage analogy.
“Embrace the marriage and the prenup, and have it all outlined, so when you are involved in an inspection with an investigator it is already clear cut and there doesn’t have to be a lot of focus on who does what because it is already outlined.”
Key Takeaways
Which entities in pharma manufacturing and supply chains are responsible for product quality according to senior FDA officials? All of them.
The Preamble to the 1978 GMPs – published 46 years ago – clearly states that adulteration provisions apply to “wholesalers, retailers, pharmacies, and hospitals as well as manufacturers.” Drug products can be adulterated in myriad ways and places in the supply chain before reaching customers.
FDA’s 2016 contract manufacturing guidance seeks to clarify the importance of delineating which parties are responsible for which activities. Using that guidance to help design quality agreements helps all GMP partners and the agency identify where responsibilities lie and where remediation needs to take place when issues arise.
Each entity in the drug supply chain is subject to FDA inspection and receipt of a 483 or warning letter. Although drug sponsors are “responsible for the quality of the drugs regardless of any agreements in place” according to FDA warning letters to sponsors, it is also clear from language in warning letters to CMOs and distributors that those entities and each party in the supply chain are “responsible for the quality of drugs” they handle “regardless of agreements in place.”
On that topic, Godwin emphasized the importance of due diligence on the part of sponsors in selecting supply chain partners and cautioned against selections based only on financial considerations and not exploring the ability of the quality units in each organization to “interact and function cohesively.”
The bottom line is that the responsibility for product quality is shared among all participants in the supply chain, and each has a legal obligation to prevent or mitigate issues anywhere in that chain can impact the patient, who expects and deserves the highest quality product. Consequently, each of those entities is subject to FDA regulation and enforcement actions.
Regular Audits: Perform due diligence on potential supply chain partners and conduct regular compliance audits to identify potential issues before they result in a warning letter. Audit both your owned and operated manufacturing sites, as well as those of your key suppliers like CMOs/CDMOs. Use of Vendor Quality Monitoring tools like Redica Systems can provide key data points like a Vendor Risk Score™.