Table of contents
Who decides how inspection results are interpreted and who decides how an inspection becomes classified as Official Action Indicated (OAI) rather than Voluntary Action Indicated (VAI) or No Action Indicated (NAI)? And how much influence does the investigator who performed the inspection have on that decision?
This topic was addressed by FDA representatives at the 2022 PDA/FDA Joint Regulatory Conference in mid-September in Washington, D.C. during the “Lunch with the Regulators” session. The process the agency uses, the players, and the role of the Consumer Safety Officer (CSO)—the agency investigator—was elucidated by CDER and Office of Regulatory Affairs (ORA) personnel in response to a question at the lunch session regarding how an OAI classification is determined.
The Process Begins with the Investigator
ORA Division of Pharmaceutical Quality Operations Director Jeffrey Meng began the discussion by explaining that the process starts with the CSOs. Investigators are trained to perform inspections and to find issues and they need to determine if those issues are significant and systemic.
FDA compliance programs and policy documents help guide investigators on how to view their findings and how to document, from an evidentiary standpoint, whether they are significant. However, the observations are just that—observations made during the inspection from the investigator’s viewpoint.
The next piece of information that comes into play is the response by the firm to the observations, potentially including documentation of any corrective actions that have been taken or are planned.
FDA staff writes a report, marrying together the investigator’s findings and the firm’s response, and that report is reviewed by agency supervisors and sent to ORA for cases where the investigator is recommending a compliance review.
A key point from Meng’s discussion is that FDA investigators make initial recommendations, after which further review takes place. If the firm’s responses are not deemed adequate or the risk not appropriately mitigated, compliance officers in ORA will generate a recommendation package for potential action that will be sent to CDER or, potentially, to the Center for Veterinary Medicine (CVM) compliance office.
The Role of the Compliance Officer
CDER Office of Compliance Senior Policy Advisor Brooke Higgins continued the explanation from her point of view. Compliance officers perform an initial evaluation of the Establishment Inspection Report (EIR)—the investigator’s documentation of the inspection—the exhibits that come with it, the recommendation memo that comes from the ORA compliance officer, and an analysis of the 483 response the firm submitted, she said.
The team leads and branch chief and division director, along with policy personnel, do an initial evaluation and come up with a recommendation that is proposed in a meeting with the office director. If there are any questions, the team can reach out to the ORA compliance officer to get information from the investigator.
Part of the evaluation, Higgins explained, includes performing a drug shortage evaluation if that is a potential issue. There are additional consultations—for example, regarding chemistry or microbiology— and involvement of any other potentially impacted center, such as CVM for an animal health product or CDRH for a combination product. That additional information is provided to the office director to help inform the final classification decision.
A Complicated Process, with Good Reason
CDER Office of Manufacturing and Product Quality Division Director Carmelo Rosa also weighed in on the process. He noted that the OAI decision involves a group effort and has been vetted through a “very complicated process—but complicated in a good way.” He also stressed the importance of 483 responses and pointed out that deficiencies in the responses can prompt warning letters to be issued.
The OAI decision, Rosa said, is based on many elements, including evidence from the inspection, the content of the 483 response, the history of the firm, the risk to patients from the deficiencies found, and whether the observations include repeats from previous inspections of the facility or another facility in the same company.
After the OAI classification is determined, another session is held to decide whether the resulting action will be a regulatory meeting or a warning letter.
Returning to the original question of how much influence the investigator performing the inspection has on the process, Rosa reiterated that the investigator does not decide whether a warning letter is issued. The investigator collects the facts, the evidence, discussions, and interactions that take place, and what occurred during the inspection, all of which is considered along with all the other elements mentioned above.
[Editor’s Note: At last year’s PDA/FDA Joint Regulatory Conference, FDA’s Carmelo Rosa examined common issues FDA investigators find concerning pharmaceutical quality systems.]
At the Intersection of Science and Law
ORA’s Meng further emphasized the collaborative nature of the process, and the reason so many players and experts are involved.
FDA is “at the intersection of science and law,” he commented. “It is a law enforcement agency that makes decisions based on science.”
Because the Code of Federal Regulations (CFR) is a set of administrative laws, violations of the CFR are violations of the law. In other words, there is a legal standard that is not being met, such as GMP adulteration or misbranding. If the agency is going to impose regulatory or judicial action, it must meet the standard that the law requires.
To that end, FDA has attorneys—regulatory counsels—who are included in the reviews and composition of warning letters, because the actions must meet legal standards.
The language is important: Agency investigators make “observations” on 483s, whereas a warning letter contains “citations,” i.e., citing violations of the law.
It is important to remember that CFR citations in a warning letter are violations of federal law and should always be taken seriously, as should any responses to the agency.
Actionable Intelligence for Monitoring Supplier Quality
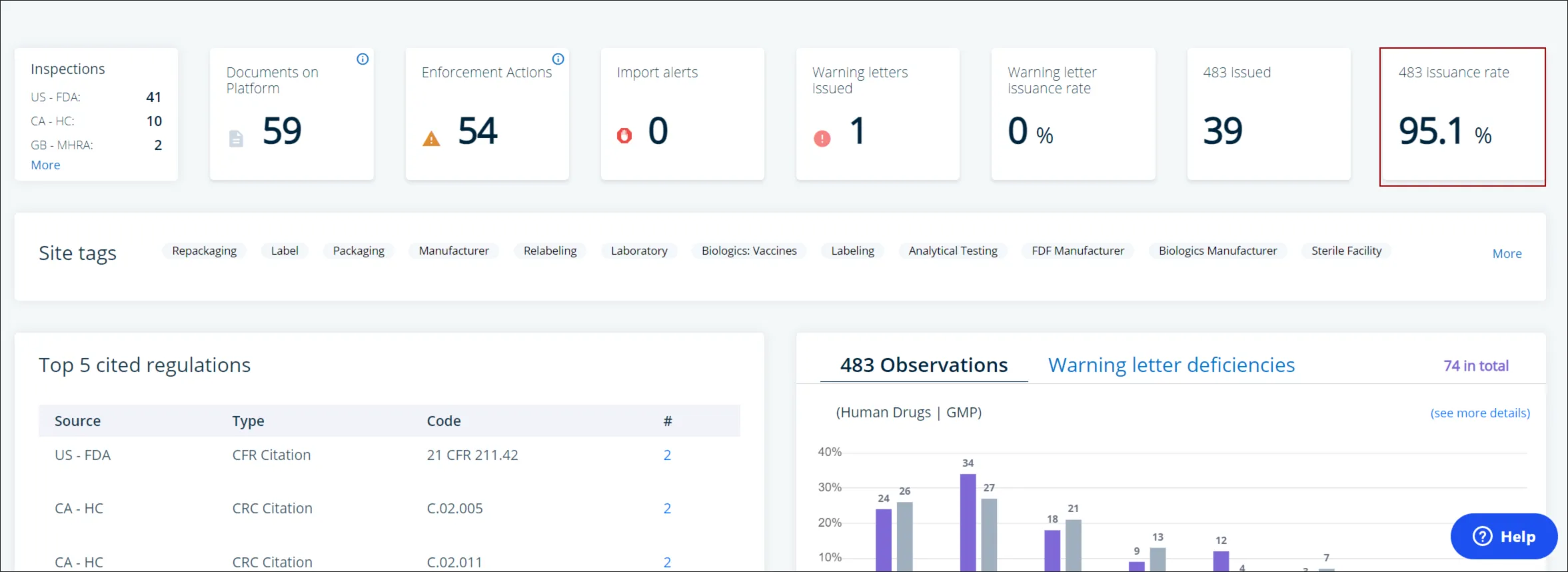
If you are considering a new contract manufacturing partnership, due diligence activities include reviewing a site or firm’s enforcement history, including 483 observations, EIRs, and 483 responses.
Redica Systems can provide you quick access to these important documents. You can also view Site and Organizational profiles for contract manufacturing organizations (CMOs). With this intelligence, you will never be in the dark about your CMO’s regulatory history and their most up-to-date inspection information. For example, one well-known CMO has a 483 issuance rate of 95.1%.
Contact us today to see how our quality and regulatory intelligence can help you identify and control risk with your critical GMP partners.