

Table of contents
ALCOA(+) – a gift from FDA’s Stan Woolen that continues to add value
The following is a guest article by ERA Sciences’ Eva Kelly. BSc Ph.D. FICI.
Author’s Note: This article, focuses the data integrity lens on original and accurate data. Also described is the ALCOA+ acronym for readers new to the data integrity topic. We hope you find the shared original ALCOA principles and best practice tips useful. Part II will discuss examples of accurate data in more detail.
In the life sciences and medtech industries, the downsides associated with unreliable data and data lacking integrity are well known and can be catastrophic—the worst, of course, being the impact on patient health resulting in harm, or even death.
There are also business impacts when data is found to be unreliable, such as delayed release to market, fines, warning letters, loss of jobs, and extensive remediation efforts to fix “poor data” processes, systems, and even people.
[Related: For more on data integrity, click here to access the full recording, including presentation slides, from a recent Redica Systems webinar.
Inspection findings from Redica Systems show 134 human drug GMP 483 observations for 2022 (at the time of going to print), following a two-year pause in onsite inspections due to the Covid-19 pandemic.
The internal and external impacts of poor data reliability and integrity have been broadly bucketed in Figure 1. Many more can be added depending on where your organization fits across the product and data lifecycle.
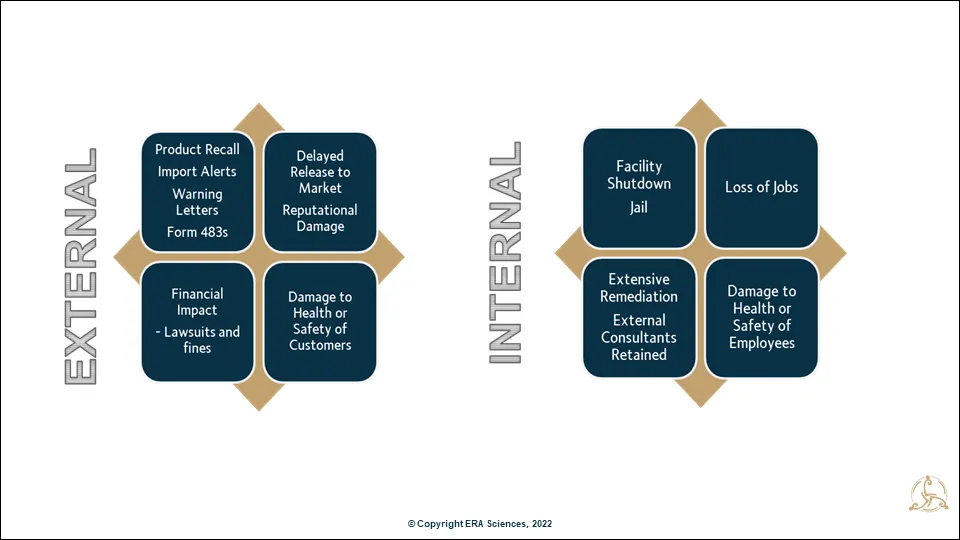
I will discuss some data integrity examples from the later commercial manufacturing phase. For starters here are examples of poor data impacting product approval regulatory decisions that by their nature delayed release to market and availability of products to patients. Obviously, protecting the patient is key, so delayed release is very much a two-edged sword.
(Just in case you are not aware, the Bioresearch Monitoring Program (BIMO), run by the U.S. FDA, oversees the conduct of onsite inspections and data audits of FDA-regulated research in support of new product development and marketing approvals.)
Typical negative data findings for NDAs and BLA inspections include:
- “Site failed to retain necessary documents”
- “Inability to locate informed consent forms and case report forms”
- “Inadequate or inaccurate record keeping including case histories, study and drug disposition records”
- “Additional data is required to support major amendments to original filings”
Recently, we have seen FDA needing more time to assess bluebird bio’s BLA, requesting additional clinical data, and pushing the review time for two of the firm’s lentiviral vector gene therapies into 2023.
Puma Biotechnology was another company that saw significant delays when its breast cancer treatment was delayed due to FDA requesting additional cancer risk safety data (animal carcinogenicity data). This pushed approval out by two years (it was finally approved in 2017).
Probably the most widely publicized involved the Novartis gene therapy product, Avexis, which nearly fell at the final post over clinical data concerns.
Could these or other data integrity issues have been avoided? Let us go back to basics!
One of the key drivers of data with integrity is ensuring data exhibits the attributes of ALCOA+/ALCOA++. I am not getting into the +/++ discussion here, though you can reach out to me by email. FDA’s Stan Woolen first used the ALCOA term back in the 1990s and probably did not realize it would become the “minimum data integrity expectation” for the next 30 years and counting!
Recommended Reading: Data Integrity 101: Why is it Important?
ALCOA+
This more extended ALCOA+ acronym now universally used describes the minimum attributes of data with integrity across the product and data lifecycle. You can look at each term to get a broad understanding of how these attributes contribute to data we can rely on to make the right decisions about product quality, safety, efficacy, and reliability (Figure 2).
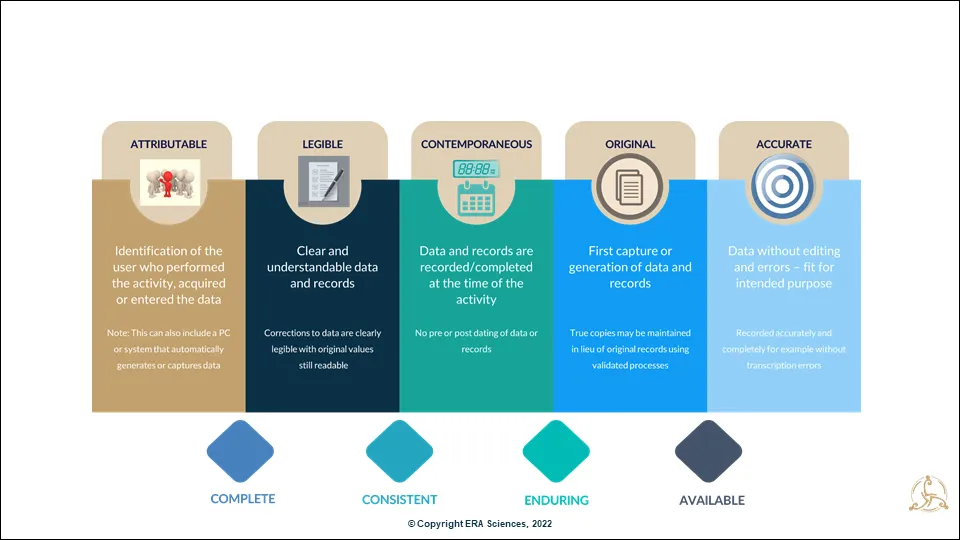
When we discuss data integrity with new and even experienced life sciences and medtech personnel, the first three “A, L, C” attributes are usually well understood; the “O” and final “A” always cause more discussion and debate, so I will focus on these for the articles in this series.
The “+” of ALCOA+ (CCEA) attributes require data to be:
- Complete – No missing or excluded data
- Consistent – Approved methods for data generation, validated, and fit-for-purpose workflows, systems, and processes
- Enduring – Media including paper and electronic solutions that are maintained, protected, and readable (interpretable) throughout the data lifecycle
- Available – Readily accessible as and when required
Recommended Reading: Data Integrity and ALCOA+
Original Data and Records
The first capture, or generation, of data and records seems quite straightforward, yet failure to correctly identify an original record continues to be an issue. If not identified correctly, then maintaining the original record throughout the required retention time is likely inadequate and flawed from the start!
Based on Redica Systems data (Figure 3) original data issues have featured frequently comprising 17% of all data integrity issues over a five-year period.
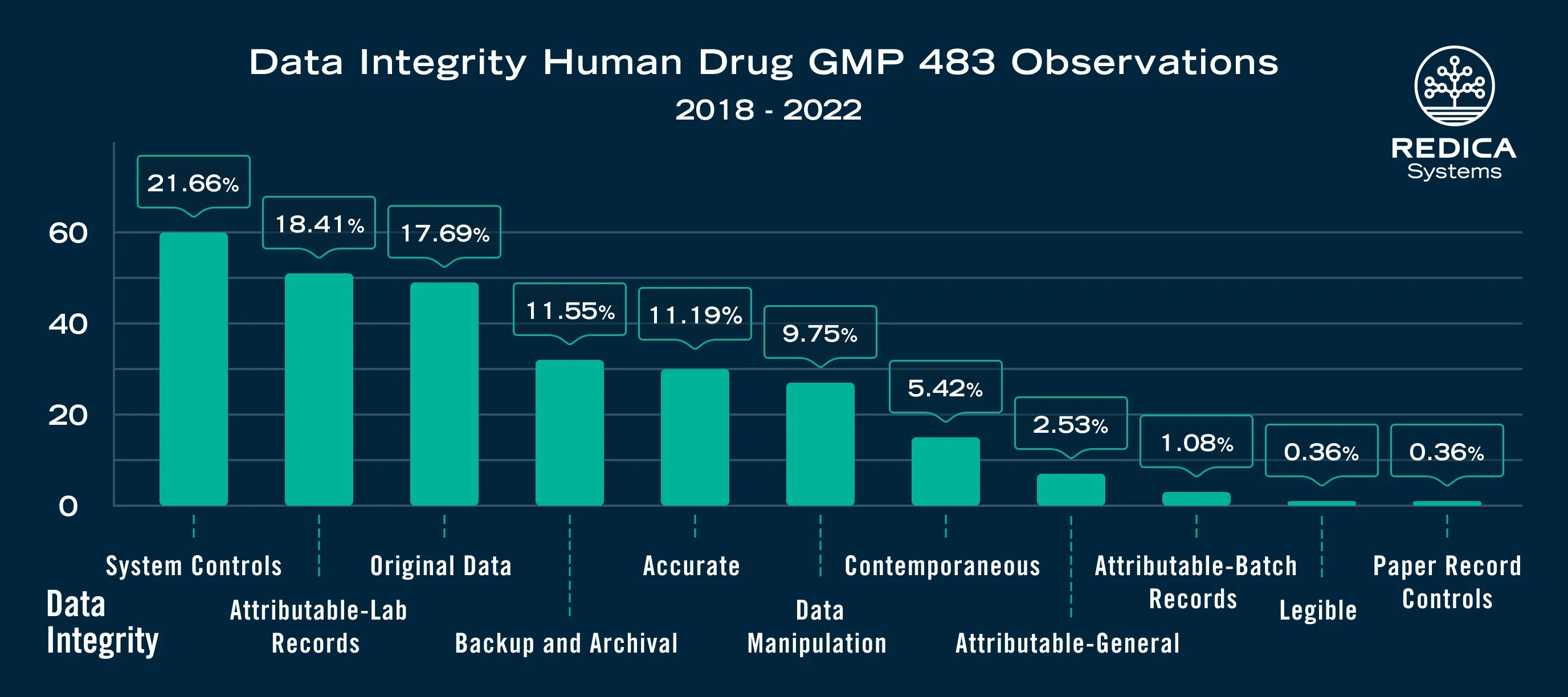
Wherever original records are generated, they need to be maintained throughout the required retention time—this does not mean that once reviewed, responsibility and control are no longer applicable. Check out this clinical site deficiency from 2020.
- “Review of the study documentation revealed that source records (e.g., informed consent documents, data record books, hospital records, and/or medical records) for subjects (b)(4), which were previously reviewed during the last inspection dated May 5, 2009, to May 29, 2009, were no longer in the subjects’ records for review.”
To add additional complexity, the use of true copies in lieu of original records also poses challenges. The 2021 PIC/S data integrity guidance very clearly calls out some minimum, but very necessary controls, including the use of controlled and/or validated processes during true copy generation, and that adequate oversight responsibilities should be in place that:
- “Verify the procedure for the generation of true copies, and ensure that the generation method is controlled appropriately.” PIC/S also describes limiting access controls to original records or true copies.
- “8.11.3 Measures should be in place to reduce the risk of deleting the wrong documents. The access rights allowing disposal of records should be controlled and limited to few persons.”
Provisioning original records, or true copies, during any regulatory inspection, aids the review and assurance of GxP-compliant activities. It will also show how good or bad record management and control actually are within a facility.
Managing Original Records
Paper Batch records. The data associated with the manufacture of the batch is directly recorded (as it happens) on a controlled, effective paper batch record, worksheet(s), and/or form(s).
- All entries are recorded by personnel manufacturing the batch or directly witnessing steps.
- Associated calculations, measurements, alarms, and deviations are directly referenced or captured on the batch record, and checks and reviews are directly recorded on the documentation.
- Additional records may be attached directly to the batch record or accurately and completely referenced.
- Effective controls, including issuance of the worksheets and forms by designated personnel help to ensure that the records are original and that photocopied, blank documents cannot be used if and when errors occur. Control-issued documents may manifest watermarks, serialization codes, stamps, and even be on colored paper. If these controls are not in place or can be circumvented, then it cannot be guaranteed this is the original record.
If any component of the batch record becomes damaged during manufacturing or review activities, a process needs to be in place to secure the original record or to generate a true copy that acts as an official and verified replacement of the original data. Lack of integrity associated with poor attribution practices, poor legibility, and non-contemporaneous recording all call into question the validity of the original record and its data.
Uncontrolled photocopying and circumvention of issuance and reconciliation controls lead to significant questions regarding the originality of the record.
Here are some examples of issues with true copies taken from recent inspections:
- “Production and laboratory personnel can copy GMP forms or print GMP forms from a shared computer drive without tracking what GMP forms are created.”
- “Document control of master and production batch records by the Quality Unit is inadequate…throughout the inspection, I observed multiple photocopy machines throughout the firm, which are not locked and available for use for all.”
- “You did not keep original records, true copies, or electronic records.”
- “The production personnel are permitted to print blank copies of GMP records, such as batch records and cleaning records. There is no control to ensure all original records are maintained.”
- “It was noted that photocopies of … calibration certificates were not verified as true copies.”
Electronic Measurement Records
The data generated from instruments may be stored directly on an instrument, printed out as a “ticket”/printout, or captured on an integrated software solution. The data could even be transcribed from the instrument readout by personnel to a paper record or system where the storage capability does not exist. Here, overwrites can happen rapidly with little control.
Understanding the instrument capability and built-in controls for data integrity should inform which of these options is the correct one. Just because an instrument or system has printout capability this does not mean that the printout is the original record.
The example I would like to discuss is Fourier Transform IR (FTIR) data and it continues to be a source of inspectional findings related to data integrity. FDA and PIC/S have made it very clear that the distinction between static and dynamic records must be understood by data owners.
Wherever original records are generated, they need to be maintained throughout the required retention time
Records that are dynamic in nature cannot be fully represented using static paper printouts or electronic PDF reports. So, original FTIR data must be available on the FTIR computerized system, either live (with suitable backups performed in case of disaster) or electronically offline in a suitable archive solution that maintains the integrity and dynamic nature of the records.
The original captured FTIR instrument data is often termed “raw data” and comprises an original record with all of its metadata. If this record is processed further on the FTIR system and generates additional result files, these are also original records.
If a printout is produced of the FTIR results and attached to other records such as a batch record, this represents a subset of the FTIR data. The original data still resides in the system. If a reviewer only examines the printout, this means the original record and all associated metadata (plus events that happened during the analysis) cannot be confirmed, e.g., additional or failed scans. Printouts do not allow us to zoom in on baselines!
Printouts and reports can also be configured to exclude data. For any type of dynamic data, original data must be understood and reviewed correctly. Although the following example describes an access control issue for FTIR data I just had to call it out—Akorn Inc. in 2018 did not exercise appropriate system controls, thus, their original data was found to be at significant risk.
The following are further examples of printouts being used to make quality decisions:
- “During data review, only final printouts of the test results are reviewed, and they are not verified against the original electronic records”
- “According to…the firm’s Quality Control laboratory, review only the printout of data generated from the High-Performance Liquid Chromatography (HPLC) systems. The original electronic data and the related audit trails are not reviewed by the Quality Control laboratory prior to the release and distribution of each batch of drug product.”
- “Currently, quality control personnel do not review system logs (audit trails) on the computers during the review process but instead review a paper printout”
What the Regulators Say
A commonly cited inspectional finding involves 21 CFR 211.68(b). Automatic, mechanical, and electronic equipment require “controls to prevent substitution or overwriting of data.” If these controls are not in place, then reviewing printed reports will not uncover discrepancies in the electronic data recording. Issues such as management processes or out-of-specification (OOS) results become invisible to the reviewer who relies on paper records rather than the electronic originals. Akorn Inc. received numerous data integrity observations in 2018 related to original data and is back in the news yet again in 2022 with repeat observations.
Solution Space
So, what can you do to ensure original data is understood and managed appropriately? Here are some tips to get started:
- Understand the required activity and data/record generation process
- Discuss the capability of instruments and systems with vendors
- Record/document instrument and system capabilities
- Apply a risk-based approach informed by sound scientific judgment, critical thinking, and accurate data to determine where paper or electronic records constitute the original record
- Be aware that hybrid original records may result from systems and instruments that cannot deliver the required data integrity; electronic systems that do not have an adequate audit trail to discern all record activities may have to be supported by additional paper-based records, e.g., logbook entries or original records
- Ensure system controls do not allow for substitution or overwriting of data
- Map the data lifecycle and determine if original data/records is being generated and managed (including oversight) appropriately
- Provide adequate oversight of manually transcribed values that cannot be stored directly and must be “first captured” in a paper or record format
Ultimately, you must identify what your original record is, and understand if the system, instrument, or paper-based process has all the necessary controls to maintain the record throughout the required retention time. Just because an instrument or system allows you to generate printouts does not mean the printout constitutes an original record. Original records need to be readily available to the right people. This means understanding and controlling access to either the live repository or the identified archival solution.

Dr. Eva Kelly is a senior consultant at ERA Sciences, with over 30 years’ experience in life sciences and med tech. She is passionate about data integrity and reliability and helping organizations build a culture of excellence associated with reliable data. ERA Sciences partners with quality committed organizations on their data journey, helping them to realize the best patient and business outcomes.
ERA Sciences would be delighted to support your data reliability and integrity efforts. Check us out at www.erasciences.com contact us at info@erasciences.com or access our learning platform Home (erasciences.com) to start learning with us today.
