

Table of contents
Redica Responds is an article series where we take common/unique questions from customers and industry events and answer them with our data. Use the following link to look at all previous Redica Responds articles.
What is the number of FDA 483 observations involving deviation investigations (inadequate, incomplete, etc.)? Is this an area that FDA investigators are increasingly looking at during human drug GMP inspections?
A robust quality management system (QMS) must include measures to analyze when deviations (departures from procedures) occurred and why, i.e., root cause, along with plans to prevent it from repeating. Not surprisingly, if FDA investigators believe that a deviation investigation was inadequate, this will be identified as a deficiency.
But how common are inspection findings involving issues with deviation investigations? Let us take a look using Redica.
From 2017 to 2022, there were 604 FDA 483 observations involving deviation investigations. Comparably, there were 739 observations involving deviation investigations during the previous five years (2012-2017).
(Keep in mind, onsite FDA inspections were down significantly due to the pandemic in 2020 and 2021.)
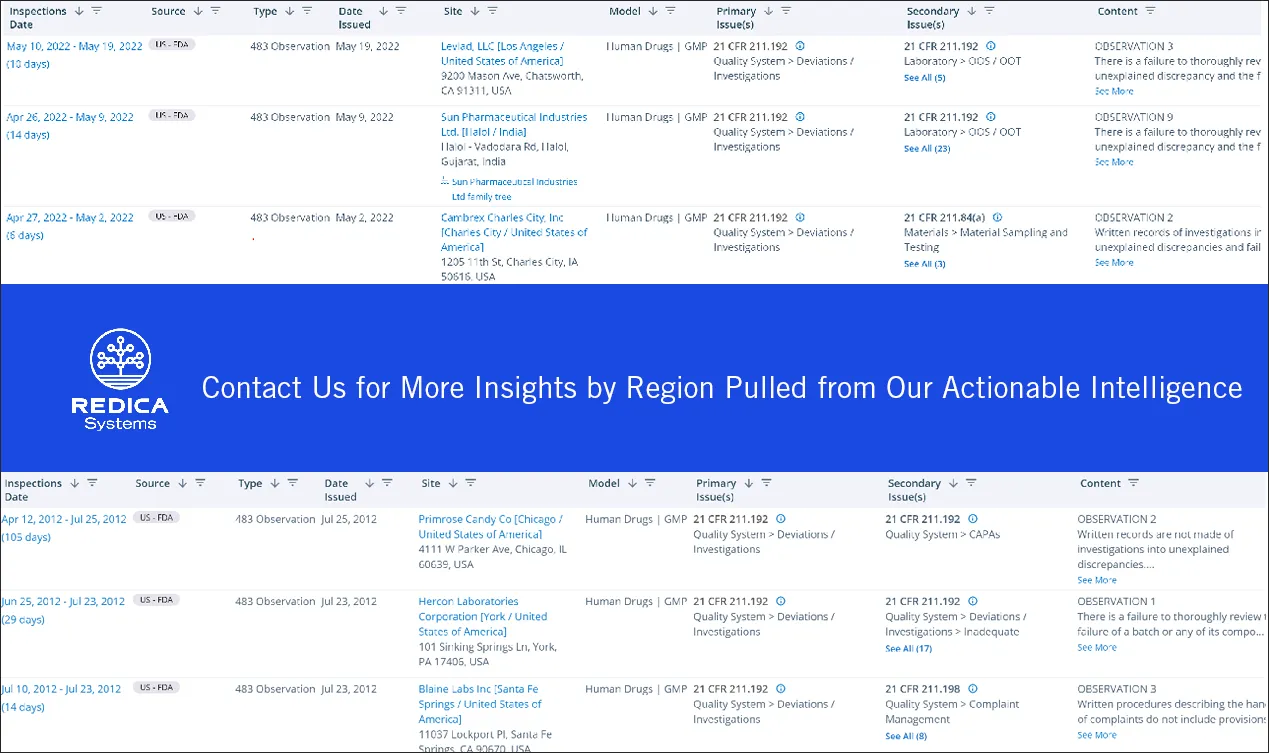
Clearly, this is an area of concern for FDA investigators. For example, during an inspection of a firm manufacturing injectable products, the investigators found failures to adequately investigate deviations involving a depyrogenation tunnel and an autoclave.
Investigators also want to see that firms outsourcing manufacturing operations work with their contract manufacturing organizations (CMOs) to ensure that the partnering manufacturer has robust deviation investigations procedures as well.
How Redica Can Help
With Redica’s actionable intelligence, you can unlock insights that help you better prepare for your next inspection and monitor vendor/CMO quality. Our Enforcement Analytics enables you to base your audits and inspection planning activity on the most comprehensive and up-to-date inspection and enforcement trends benchmarked to the industry.
Contact us today to learn more about how we can help you stay on top of the latest inspection findings involving deviation investigations and other issues.
