

Table of contents
FDA-regulated firms face an enormous challenge keeping up with agency priorities and expectations—especially companies that produce older products within an ever-changing regulatory landscape. While firms continue to manufacture these older products to serve their customers and protect their brand, there are many potential quality compliance issues that can arise.
The products in question are known as “legacy products.” According to a paper published by Pew Charitable Trusts and ISPE in January 2017 on drug shortages¹, a legacy product is a “previously approved and marketed drug, typically developed 10 to 20 years ago. Such products typically have multiple competitors on the market and are low margin.”
Although the definition says, “typically developed 10 to 20 years ago,” there are legacy products that are much older than that. There are products that have been on the market since the forties, fifties, sixties, and seventies. The seventies are when modern GMPs began. 21 CFR 210 and 211 were greatly expanded and finalized in 1978, and a lot of things have changed since then.
Prior to the late seventies, drug candidates were selected using quality parameters of the API, like taste and odor. Stability studies were generally done at lab scale and were only run for weeks or months. Scaling up to industrial scale was often done by adjusting production equipment in a manufacturing plant, which may or may not be documented. Very little attention was paid to possible interactions between the active ingredient and the excipients or to degradation pathways of the active ingredient. Hold times usually were not thought about much.
Prior to the GMPs in 1978, drug product quality and sterility were based solely on finished product testing. Terms we are very familiar with today such as “validation” and “quality control unit” were not in the pharma lexicon until FDA introduced major changes to the GMPs 41 years ago.
[Note: You can have the information presented here for your reference — download the slide deck and webinar presentation here!]
The Pharmaceutical Quality System
Clearly, a company needs to have demonstrated conformance with the rules that were applicable when a product was approved for marketing. However, a lot of changes have occurred over time. We have seen rapid changes just over the last decade—over the last five years, even, to manufacturing technologies, lab analysis, and detection methodologies and abilities, and to what health authorities expect. In some cases, companies will be expected to conform to rules that went into effect after their product was approved.
A major change that took place in the early 2000s was the introduction of the “pharmaceutical quality system” (PQS). It was originally proposed in an FDA initiative, the Pharmaceutical CGMP Initiative for the 21st Century—a Risk Based Approach², and formalized six years later in ICH Q10—The Pharmaceutical Quality System.³
The International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use (ICH) has membership from the US, EU, and Japan, including from both industry and regulatory agencies. When ICH guidance documents are written, they are reviewed through the three agencies, finalized, and then adopted by the three agencies. Note that this is guidance—it is not regulation, so it is not something that companies will generally be directly cited directly against. But it is guidance and it does create expectations in the US, the EU, and Japan.
ICH Q10 describes one comprehensive model for an effective pharmaceutical quality system. It is based on the International Standards Organization (ISO) quality concepts, references good manufacturing practices, and complements ICH Q8 on Pharmaceutical Development⁴ from 2009 and ICH Q9 on Quality Risk Management⁵ from 2005. Guidance from that suite of three documents is integral to creating an effective pharmaceutical quality system.
Within ICH Q10, the term “continual improvement” is used 25 times in the 21-page document. There is an expectation for continual improvement of products and processes, which specifically applies to both new and existing products. This is important to note as continual improvement can be challenging in the context of legacy products.
Other potential GMP challenges can arise due to changes over the years in expectations, or new expectations resulting from:
- FDA inspection models
- CPGMs
- ICH Q7
- ICH Q3D
- FDAAA
- ICH Q3C
- Quality agreements
- A focus on data integrity
- Process validation
FDA Inspection Models
Regarding changes to FDA inspection models, one might say that legacy products, by definition, have been around a while, and have probably been inspected many times. If that is the case, then why be concerned now about agency inspections? There are a few reasons.
One is FDA’s Program Alignment initiative.⁶ FDA has created distinct product-based, vertically-integrated regulatory programs with trained investigators. That means that it now has investigators who are specializing by FDA regulated product type.
FDA inspects vaccine manufacturers, blood manufacturers, blood banks, food processing facilities, dairy farms, animal feed processors, and compounding pharmacies. In the past, an FDA investigator could have inspected a blood bank one week and a dairy farm the next week, and then a week or two later a sterile drug manufacturing plant. How much did the investigator really know about each of the various facilities? That is variable.
What has changed is that now each investigator is going to have an area of expertise. If a company manufactures sterile products, the person who comes in to inspect is going to be someone who has expertise in sterile products, or in solid oral dosage forms, for example, if that is the type of facility being inspected.
Program alignment is phasing in. It began in mid-2017. Companies are going to see more informed FDA investigators coming into their facility—investigators who better understand their products and processes, which will result in more rigorous inspections.
A second major change in FDA inspection models is the New Inspection Protocol Project (NIPP).⁷ It consists in part of a set of expert questions that are used during the inspection. It was piloted in sterile drug products and began being applied in other product type inspections in September 2019.
NIPP shifts focus from basic compliance with good manufacturing practices to looking for more of a constant state of quality. Investigators will be looking for quality systems that support variability reduction and continual improvement, like in ICH Q10. They are also looking at each company’s quality culture. These inspections are going to be different than facilities have experienced in the past.
The inspections will continue to use a quality systems approach. The agency has made it clear that the protocols they are using will not be made public, but there are some aspects of the program and ways it might be used that have been elucidated in public presentations by agency personnel.
The surveillance inspections will continue to focus on the six quality systems, but these systems will be broken down into 29 elements, and each of those is going to be scored separately. Each has six performance levels (see Figure 1).
Figure 1
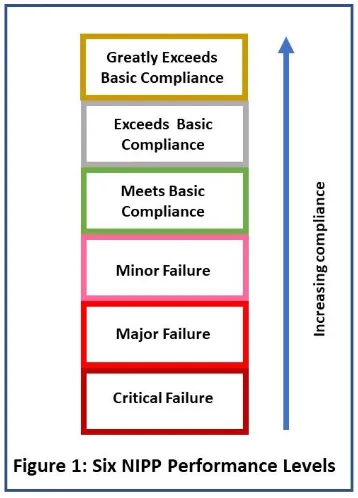
Figure 1 did not come from FDA. It is based on a verbal FDA presentation that listed the six different levels of compliance for each of the elements and the language used.
Although the elements have not been disclosed, it has been implied that they are areas that are typically covered during inspections. It has also been implied that the resulting scores are going to be semi-quantitative to allow comparison across sites.
This approach will not impact the official inspection outcome. When an FDA investigator completes an inspection, if there are no findings, it is NAI, No Action Indicated. If there are any findings, then it gets categorized at the Center as either Voluntary Action Indicated (VAI) or Official Action Indicated (OAI). Those designations consider not only what happened during that inspection, but also historical and other information. So, the use of the NIPP methodology is not going to impact the overall categorization.
Also keep in mind that FDA investigators vary a lot in their approach, in their demeanor, and in their expectations. We have all experienced investigators who are not as easy to work with as some others or some who tend to march to a different drummer or maybe more career-minded, which is the same as in any group of professionals. Different personalities and approaches add to the variability of the inspection conduct and outcome. (Note: for more information on a specific investigator’s inspection history, click here).

And lastly, perhaps the last time an investigator inspected a facility it was for a pre-approval inspection. So even though the site was also manufacturing a legacy product, the investigator did not really look at it. Or maybe he or she chose during a routine surveillance inspection to look more closely at new products rather than one that the company has been manufacturing for years.
In other words, it is possible that it may have been a while since a legacy product was inspected. But now, with program alignment, the next investigator will understand more about the product type that he or she is inspecting.
Compliance Program Guidance Manuals (CPGMs)
Next are Compliance Program Guidance Manuals.⁸ CPGMs are documents that the agency puts together for their investigators to use. They are quite informative. It would be a good idea for companies to look at the CPGMs that cover their product type.
The CPGM for Sterile Drug Products (7356-002A)⁹, which was last revised in 2016, says “pay special attention to legacy products.” The phrase “pay special attention to legacy products” will be more meaningful to an investigator who has experience in sterile drug manufacturing than it would be to someone who was not an expert (see Figure 2 for an example title page from a CPGM).
Figure 2

Another CPGM (7356.002F) covers Active Pharmaceutical Ingredient (API) Process Inspections.¹⁰ When FDA inspects an API facility, it cannot cite 21 CFR 210 and 211, because the CFR regulations are only applicable to finished dosage products. What the agency uses to guide them for Active Pharmaceutical Ingredients is ICH Q7.¹¹ The CPGM employs ICH Q7 as a quality standard and uses it to establish compliance with the statutory requirements of the FD&C Act, the legal basis for FDA taking compliance action. It notes that “firms that follow ICH Q7 generally will be considered to comply with the statutory requirement.”
The CPGM for APIs specifically directs the investigator to review the impurity profile for each API process and compare the impurity profile to that which was submitted in the application or in the Drug Master File. It goes on to say that the investigator should be familiar with USP <1086> on “Impurities in Drug Substances and Drug Product” and if an impurity profile has not been filed to CDER to review the guidance on established impurity profiles in ICH Q3A and Q3C.
For a company manufacturing a legacy API, it would be good to review ICH Q7 to ensure the API meets Q7 expectations.
ICH Q3D on Elemental Impurities, FDAAA, and Q3C on Residual Solvents
ICH Q3D¹² is a guidance on Elemental Impurities in drug products. The scope is new, finished drug products, and new drug products containing existing drug substances. Keep in mind that protection of public health and safety is FDA’s mandate, regardless of when the product was approved. Even though it is only officially applicable to new drug products, it does list impurities that have been determined to be a serious safety issue, that probably were not on the radar screen of whoever developed the legacy product at the time, so it is something worth considering. And the list is revised periodically with new elements added.
The FDA Amendments Act¹³, known as FDAAA, was recently employed regarding an elemental impurity concern. FDAAA is a tool that Congress provided the agency in 2008 that was not used much until recently. It allows FDA to require additional post-market studies for prescription drugs and biologics, including risk assessments if the agency has reason to believe that there is some sort of risk.
In 2017, FDA became aware of the potential for lead in talc. Talc is a mined product. FDA assigned the Lymol Medical Corporation to perform a risk assessment on lead in its sterile talc powder product. The product has been on the market since 2003.
In February 2018, FDA notified Lymol that it was required to conduct a “Post-Marketing Risk Assessment,” PMR 3332-1, and gave Lymol 90 days to complete the risk assessment. The company did not respond. The following August, FDA notified Lymol in a “Missed PMR Milestone Letter” of its failure to comply with the final report submission date, and it gave the company 30 days to submit an explanation.
In September of 2018, FDA issued a “Failure to Respond Letter” and gave the company 30 days to respond. When it did not, the matter was referred to the Office of Compliance. A Warning Letter¹⁴ was issued in January 2019. The Warning Letter was issued because the firm failed to comply with the timetable for completion of the Post-Market Risk Assessment and failed to demonstrate good cause for non-compliance.
This is an example of a case where FDA became aware of a potential issue or concern and asked a company manufacturing a legacy product to assess that risk.
Related to that, Johnson & Johnson has been the target of lawsuits regarding alleged asbestos in legacy talc products pointing to the link between asbestos exposure and mesothelioma. J&J was ordered to pay $29 million in one lawsuit in March 2019. There have been other lawsuits that have been thrown out or turned down, and a number are still pending. Remember that this is not peer-reviewed science, but a jury trial.
ICH Q3C¹⁵ on Residual Solvents covers acceptable levels of various solvents in drug products and is updated periodically. It is probably a good idea for a company to look at the list of solvents to see if it is using any of them.
FDA Quality Agreement Guidance
FDA’s Quality Agreement Guidance¹⁶ was published in 2016. The guidance provides expectations for agreements between a product owner and a contract manufacturing facility to clearly define in detail what the GMP related roles are for each partner. The GMP responsibilities are required to be defined in detail.
Whether a firm is engaging a Contract Manufacturing Organization (CMO) to manufacture or the firm is a CMO, it is expected that quality agreements are in place between the two parties. While it is guidance and not regulation, it has been cited in several warning letters.
In 2018, at least 20 warning letters were issued to firms who were CMOs or contract laboratories, or to firms, FDA felt did not exercise appropriate controls over their contracted operation. This continues the focus that began after the 2016 Guidance was released.
Here is an excerpt from a warning letter to VitaPurity Corporation¹⁷ (see Figure 3). There appears to be a lot of assuming and no verifying.
Figure 3

In another letter citing FDA’s Quality Agreement Guidance, in June 2017 a CMO that was making oral pharmaceutical solutions for another company using the same equipment is used to make toxic car washes and waxes. ChemRite CoPac¹⁸ in Wisconsin was cited for, among other things, the lack of a quality agreement with the firm that it was making the solutions for (Sage Products).
The FDA sent a similar warning letter to Sage Products¹⁹ in July 2017 for the lax oversight of a contractor (ChemRite CoPac) that was producing the oral solutions. This is what one FDA investigator calls a “twofer”—two warning letters from one inspection. One was to the product owner and one to the CMO.
In each letter, FDA states that contractors are extensions of the manufacturer and manufacturers are required to ensure that their drugs are made in accordance with FDA regulations. In other words, a company could be a product owner or a CMO and get a warning letter from someone else’s inspection, from a CMO or vice-versa, citing issues with the quality agreement. This could happen for any product, including a legacy product that is being manufactured by a CMO.
Data Integrity
In 2016, FDA published a draft guidance on Data Integrity, which was released as a final guidance in 2018.²⁰ The predicate rule was promulgated in 1997. Approximately 80% of all warning letters in 2015 and 2016 included a data integrity component. And about 70% of the published Eudra GMP Non-Compliance Reports have similar shortcomings. In 2017, 68% of all FDA warning letters, drug GMP warning letters, cited data integrity. It is a strong current focus for all products, not just for legacy products.
For legacy products, it may be difficult to locate old documentation—for example, a development history report. It would be a good idea for a company manufacturing a legacy product to pull together any development history that is available, take a critical look at it, and have some sort of story to tell.
For a lot of legacy products, especially solid oral dose products that have been manufactured for decades, oftentimes the know-how to make the product was not so much procedure-based as manufacturing plant-based or operator-based.
One example is an operator, let’s call him Kenny, and everyone knows that Kenny knows how to run the press and make perfect tablets. That is great for the company and for Kenny. But then Kenny retires, and tablet problems begin. That certainly has happened and there may be little or no documentation on how the process was optimized and the changes that have taken place.
It is advisable from a data integrity standpoint to pay special attention to the analytical labs. Analytical labs are where a lot of the data integrity inspection citations are found. One common finding is chromatography data system audit trails disabled. A chromatography system should have an audit trail function, which means that everything that takes place on that piece of equipment is recorded and logged.
If the audit trail is disabled, then someone could be doing test injections or deleting injections—there is no way to know. If lab personnel can enable and disable an audit trail with no record that has taken place, that is an issue. It is also an issue if analysts have access to change data or are sharing passwords. Each of those situations makes the numbers that come out of the lab suspect and will be cited.
Process Validation
Process validation is fundamental to pharmaceutical processes. While there is no specific requirement for validating legacy product manufacturing processes, in FDA’s 2011 Guidance, legacy products are specifically mentioned (see Figure 4).
Figure 4

Because legacy products have been around a long time, the firms manufacturing them should have a lot of historical data. FDA’s Process Validation guidance has been in place since 1987, but it had a major revision in 2011.
If a company has a process that is in a state of control, and if it has a decent process capability (CPK)—for example, 1.2 to 2.0—then performing a Stage 3, “continued process verification,” is all that would be necessary and should not be time-consuming or challenging. If the process capability is low, then it could end up being a lot more work, beginning back at Stage 1 or 2.
Although performing process validation on a legacy product is not a requirement, it is important to note that FDA did specifically mention legacy products and how they can take advantage of the guidance. It appears to be a clue there that there is some sort of expectation or recommendation.
ISPE has some good resources for companies who decide to go down the process validation path, including a discussion paper.²¹
Combination Products are a Special Case
Any combination of drugs, biologics, or devices is considered a combination product by FDA. In the past companies followed the regulations for the individual components—for example, 21 CFR 210 and 211 for drugs, Part 820 for devices, Part 600-680 where GMPs are applicable for biologics, and Part 1271 for human cell and tissue products.
In 2013, FDA issued GMP Part 4,²² which was intended to clarify and explain the application of the various requirements to combination products. It included no new GMP requirements. A draft guidance issued in 2015 and finalized in 2017²³ explains that how existing rules apply to combination products depends on the specific circumstances under which the combination product is produced, packaged, and marketed.
If the product includes a medical device component, then a design history file is expected. That is part of a regulation that was introduced in 1990 when Congress passed the Safe Medical Devices Act.²⁴ It established standards for medical devices that can cause or contribute to death, illness, or injury of a patient.
If a company has an older product with no design history file, it will still have basic information on the product—especially if the product was patented. One example would be information on a pre-filled syringe, which can be captured to comply with design controls for existing products. It does not have to be as extensive as would be expected for a product in development now.
While documented design controls may not exist for legacy products, FDA does not expect sponsors to recreate information that did not exist previously. The preamble to the GMP rule allows for flexibility in design controls. The agency has indicated that many sponsors have information that would address some aspects of design control. And the agency can show flexibility with existing products and especially older products.
In the January 2017 combination products final guidance, FDA notes, “Although products developed prior to promulgation of part 4 are frequently termed ‘legacy’ combination products, FDA deliberately does not use this terminology in this guidance because it could be inappropriately interpreted to indicate that products developed prior to promulgation of part 4 are therefore subject to fewer CGMP obligations than products developed after promulgation of part 4.”
In 2016 at the PDA/FDA Joint Regulatory Conference, FDA Office of Combination Products (OCP) Senior Program Manager Melissa Burns said that for legacy products the agency does not expect sponsors to go back and reinvent. OCP is not trying to add unnecessary documentation and work.
At the same conference, there was a case study from Medimmune for its FluMist product, which is a legacy product. The company decided to retrospectively determine where it stood relative to design controls on the FluMist drug-device combination product. The biggest gap that it identified was a user-focused risk assessment, so one was performed.
Ultimately, Medimmune assembled a package of data that could be shared upon inspection that constituted a design history file. It was certainly different than a DHF developed de novo for a new product, but it did meet the intent of the regulations. Also, the presenter suggested that information for legacy drugs and devices for a combination product be developed separately and brought together with a cover page that includes risk and design controls for the two constituents.
¹ Pew Charitable Trusts and ISPE report, Drug Shortages
² Pharmaceutical CGMP Initiative for the 21st Century – a Risk Based Approach
³ ICH Q10 – The Pharmaceutical Quality System
⁴ ICH Q8 – Pharmaceutical Development
⁵ ICH Q9 – Quality Risk Management
⁷ FDA NIPP
⁸ Compliance Program Guidance Manuals
⁹ FDA CPGM 7356.002A, Sterile Drug Process Inspections
¹⁰ FDA CPGM 7356.002F, Active Pharmaceutical Ingredient (API) Process Inspection
¹¹ ICH Q7, Good Manufacturing Practice Guidance for Active Pharmaceutical Ingredients
¹² ICH Q3D Guideline on Elemental Impurities
¹³ FDAAA
¹⁴ Warning Letter to Lymol Medical Corp.
¹⁵ ICH Q3C
¹⁶ Contract Manufacturing Arrangements for Drugs: Quality Agreements Guidance for Industry
¹⁷ Warning Letter to VitaPurity Corporation
¹⁸ Warning Letter to ChemRite CoPac
¹⁹ Warning Letter to Sage Products
²⁰ FDA Data Integrity Guidance
²¹ ISPE Process Validation Lifecycle Implementation for Existing Products
²² GMP Part 4 (combination products)
²³ CGMP Requirements for Combination Products – Final Guidance
