Table of contents
At the PDA/FDA Joint Regulatory Conference 2024 held in Washington, DC, September 9-11, 2024, FDA Center for Drug Evaluation and Research (CDER) Office of Pharmaceutical Quality (OPQ) Office of Pharmaceutical Manufacturing Assessment (OPMA) Senior Pharmaceutical Quality Assessor Dr. Madu Dharmasena provided her insights on CDER’s regulation of biologics products.
In addition to its responsibility for drugs, CDER is responsible for regulating and inspecting certain biologics. CDER-regulated biologic products include enzymes, monoclonal antibody, antibody conjugates, fusion proteins, growth factors, cytokines and botulinum toxins.
Her review of inspection trends showed that total regulatory actions on Biologics License Application (BLAs) have been increasing in recent years. She pointed to an overall increase in Complete Response Letters (CRLs) after inspections for many reasons, with the greatest increase due to facility deficiencies. A CRL is sent by the agency to the drug applicant to inform it that the drug application is not ready for approval.
In her presentation, Dharmasena covered:
- Types of CDER-led inspections
- Inspection objectives
- How facilities are selected for inspection
- Alternatives to inspections and communication of results
- Common deficiencies found in inspections and trends over time
Types of Inspections
Dharmasena listed the four types of CDER-led inspections of biologics:
- Pre-licensing inspections (PLIs) for biologic products and pre-approval inspections (PAIs) for small molecules and BLA supplements
- Post-approval inspections
- Surveillance inspections, and
- For cause inspections
CDER performs PLIs in support of original BLAs and resubmissions and PAIs in support of prior approval supplements for the addition of new manufacturing facilities.
BLA approval includes licensing of the product, facilities, and the manufacturing process. The manufacturing process needs to be validated and in operation during the inspection.
The pre-licensing inspections and pre-approval inspections effectively tie the integrity of the data and information provided in the application to data generated at the facilities. Inspections completed with sufficient time before the user fee date can incorporate all the findings into the application.
The CDER biologics reviewer pointed to some of the key regulations governing CDER inspections of biologics (Figure 1, Regulations for BLA Inspections).

Figure 1 | Regulations for BLA Inspections
Primary Inspection Objectives
Dharmasena said the inspections have three primary objectives.
- Readiness for commercial manufacturing, “where we verify that the facility named or referenced in the application can perform the proposed function in conformance with good manufacturing practice requirements.”
- Conformance to the application, “where we determine that the facility is following the commitments made in the application.”
- Data integrity audit, “where we confirm the data submitted in the application are accurate and complete.”
How Are Facilities Selected for Inspection?
Dharmasena explained that CDER uses a risk-based approach to determine the need for a PLI or PAI. “We use the information in the application as well as the information available to us at FDA to make the determination. Some of the questions that we ask are:
- “Does the facility and the proposed manufacturing areas have biological inspection history via FDA or mutual recognition agreement?
- “Is there a gap in recent inspection history?
- “Are there any unresolved potential GMP concerns in areas related to the process in the BLA?
- “Are there any systemic problems such as QC/QA oversight?
- “Is the site experienced with similar equipment and processes?
- “Are there any product specific concerns identified from the BLA review itself?”
Alternative Tools Supplement FDA’s Toolbox
Various tools in addition to on-site inspections can be used to satisfy the inspection objectives – for example, 704(a)(4) record requests, Remote Interactive Evaluations (RIE), or inspection waivers based on inspection reports from other health authorities that FDA has a Mutual Recognition Agreement (MRA) with.
“We started using alternative tools during the pandemic, which include a document request under section 704(a)(4) of the Federal Food, Drug, and Cosmetic Act. They are not mutually exclusive,” Dharmasena pointed out. “Sometimes we perform a 704 document request prior to an inspection so that we can shorten the length of the inspection and to have the inspection more targeted.”
Since these alternative tools are not considered inspections, no 483 is issued. “However, we do issue an observation letter to enhance transparency and to provide a list of unresolved issues. For on-site inspections, we do issue a 483 after the inspection and the inspection team makes an initial field recommendation.”
“If we find many, many deficiencies, then a Withhold status is recommended. We communicate with the sponsor or the applicant that the inspection team has identified deficiencies and satisfactory responses from the facility are needed before this application can be approved. Then the firm has three weeks or 15 business days to respond to the 483 observations.”
“When these responses are received, we perform the compliance review. The compliance review includes reviewing the responses and providing the final recommendation. If the initial recommendation is Withhold, and if the firm has satisfactory responses to these observations, we can downgrade the recommendation to Voluntary Action Indicated (VAI). However, if the final recommendation is Withhold, we issue a post action letter after the complete response action.”
Common Inspection Deficiencies and Trends
“The number of BLAs that we have been receiving has been increasing,” Dharmasena said. “Total actions on BLAs have been increasing recently, with a concurrent increase in CRs for all reasons.” The greatest increase has been in facility deficiencies (Figure 2, Complete Response Letter Deficiency Trends).
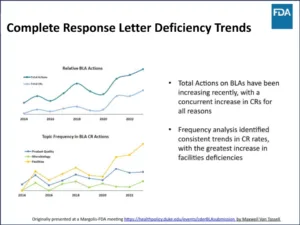
Figure 2 | Complete Response Letter Deficiency Trends
Issues are found in all six inspection systems – the quality system, facility and equipment, materials, production, packaging and labeling, and laboratory control systems.
“Usually, the facility “Withhold” recommendation results from multiple issues. Common categories of outstanding deficiencies appearing in post-action letter include:
- environmental monitoring and personnel monitoring
- data integrity
- process control and validation
- equipment cleaning and cross-contamination
- visual inspection
- laboratory SOPs and investigations
- quality agreements
- disinfectant efficacy and facility sanitization
- equipment maintenance, and
- personnel monitoring programs.”
CDER also performs biologic drug substance inspections.
“The biologic drug substance manufactured through a biotechnology process refers to the use of cells or organisms typically to produce a drug substance. A drug substance produced by a biotech process normally consists of high molecular weight substances such as proteins and polypeptides.”
“Production of drug substances from cell culture or fermentation includes biological processes such as cultivation of cells or extraction or purification of material from living organisms. Such a process is prone to contamination. Appropriate controls should be established at all stages of the manufacturing process to ensure drug substance quality. Appropriate, qualified, and validated facility equipment, process design, and environmental controls should be used to minimize the risk of contamination.”
In Part 2 of this article on Dr. Dharmasena’s presentation, we will review the case studies she presented on inspection findings.