Table of contents
*Updated April 26, 2023*
China’s huge pharmaceutical market has long attracted the attention of international drug companies. But ferreting out how to gain access to the market and conduct clinical trials there has been elusive.
China’s government recognized some time ago the benefits large pharma companies could bring to the country and its people and has slowly begun examining how to enable Western companies to gain access to the Chinese market.
[Related: Stay on top of regulatory surveillance within the Asia-Pacific region. Contact us today to see how our regulatory surveillance can help you stay on top of global regulatory developments.]
In the past few years, China’s government has developed and approved many policies and measures that encourage innovative and global drug development and significantly improved review timelines and processes for gaining new drug and clinical trial approvals.
Included in the recent reforms is a staged implementation of ICH guidelines aimed at aligning China’s requirements with international standards.
At PharmaLink 2021 co-sponsored by FDA and Xavier Health, Pfizer Senior Director and Head of Global CMC for China for new and marketed products Xiaoping Cao provided an in-depth look at the latest legislation and regulation in China and insight on how pharma companies can navigate its revised regulatory process.
Recent Legislation And Developments
Cao explained that there are multiple layers making up China’s drug regulatory framework (Figure 1). At the apex is the law—legislation that includes, for example, the Drug Administration Law (DAL) and the Vaccine Administration Law (VAL). The most recent revisions were effective December 1, 2019.\

Under this level there are regulations. One of the most often referred to is the Drug Registration Regulation (DRR), which was recently revised, and became effective in July 2020.
Below this layer is the China Pharmacopeia, usually referred to as the ChP. Every five years the ChP gets updated. The most recent version is called ChP 2020. It was effective on December 30, 2020. [Editor’s Note: See “Chinese Pharmacopoeia 2020 Includes Updated Chapters in Key Areas” for more about this edition of the Chinese Pharmacopoeia.]
There are also many detailed technical guidelines—for example, for clinical trials, new drug applications, and post approval variation guidelines.
“We often refer to the China Regulatory Reform that began in 2015 with notice number 44,” Cao said. “That was a milestone that started the regulatory reform (Figure 2). In 2017, China joined ICH. I also mentioned DAL, VAL, and the revised DAL. And after that, there are many CMC-related guidelines.”
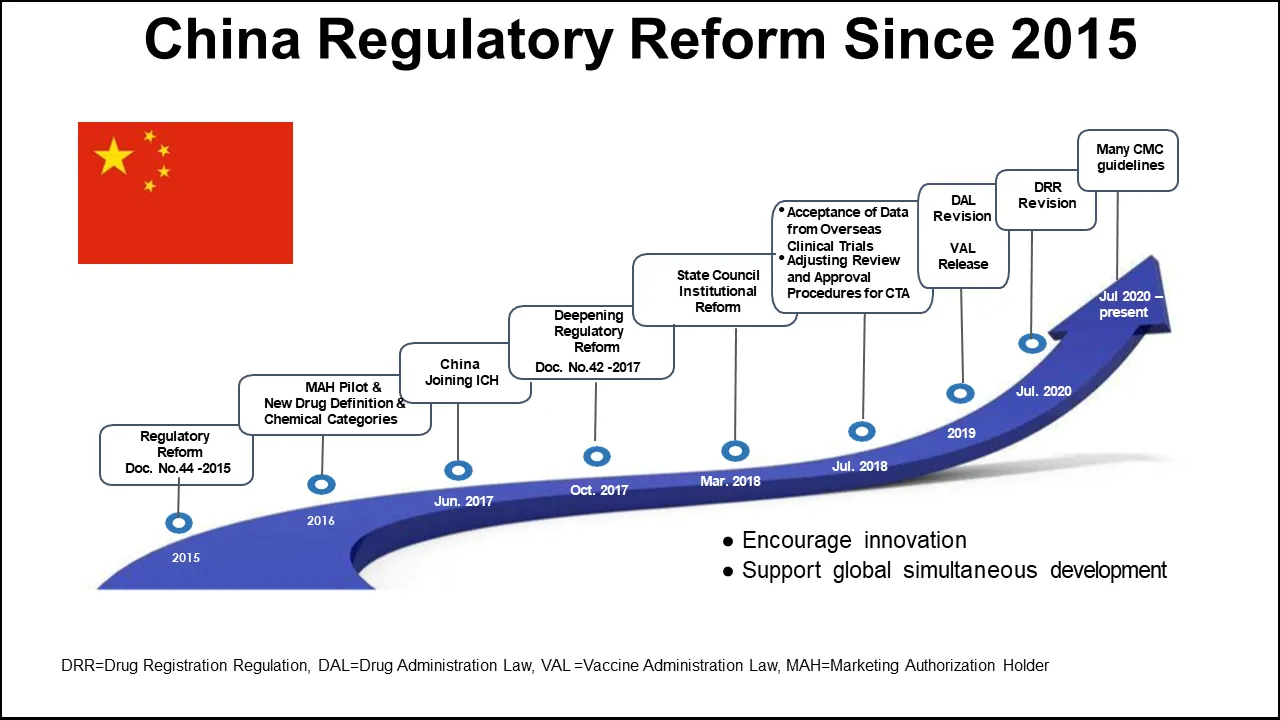
“All this comes nicely together to encourage innovative drug development and support of global simultaneous development allowing China to join global studies,” he commented.
A very substantial change is the approval time for a clinical trial application (CTA). In the past, it took two to three years for CTA approval. And now, by default, it is a 60 working day “silent approval,” meaning if no response is received in 60 working days the application is approved.
Another important point is global simultaneous development—many multinational companies are coming to China earlier and earlier. In the past, many drug trials came to China only after a major market approval. They would join in Phase 3. Now they often join in Phase 2 or even earlier. Many revisions and policies now encourage innovative drug development. The timelines and processes have been “greatly improved.”
Cao summarized some of the ICH-related guideline implementation progress in China (Figure 3). It is based on tiers. For example, Tier 1 guidelines Q1 and Q7 were implemented immediately after China joined ICH in 2017.

Some other guidelines were implemented later and others are planned. Although still in the planning stage, and not implemented yet, China has indicated a “great interest” in the Q12 guideline.
There are multiple accelerated drug approval pathways that have been defined, such as breakthrough, conditional approval, priority review, and special approval (Figure 4). Those pathways greatly facilitate expedited drug approval, especially for drugs with unmet medical needs.

“If you look at some of the criteria, you can see there are many similarities with those of the U.S. FDA,” Cao pointed out. “The benefit to the agency, to the company, and mostly to the patients, will be getting innovative drugs to patients earlier.”
The Chinese Pharmacopoeia, ChP 2020, implemented at the end of December 2020, consists of 4 volumes, which comprise general notices, monographs, and general technical requirements of Traditional Chinese Medicine, chemical drugs, biologics, and excipients.
All drugs manufactured and/or marketed in China must conform to relevant technical requirements in the latest edition of the ChP.
Since 2019, China has been revamping its Drug Administration Law (DAL) with a flurry of new requirements across GXP areas and topics, from the more general topics like Drug Innovation all the way through the specifics like Drug Traceability System.
The industry has had difficulty following up on all document iterations, with limited windows to comment before it becomes in effect.
One key branch is strengthening the Marketing Authorization Holders (MAHs) GXP responsibilities to ensure the Safety and Quality of Drugs. After a short consultation period, the new requirements have been finalized and came into force on 1 March 2023. In the same vein, China NMPA also published its expectations regarding the Medical Devices Quality Management System (QMS) piece.
China-Specific Requirements
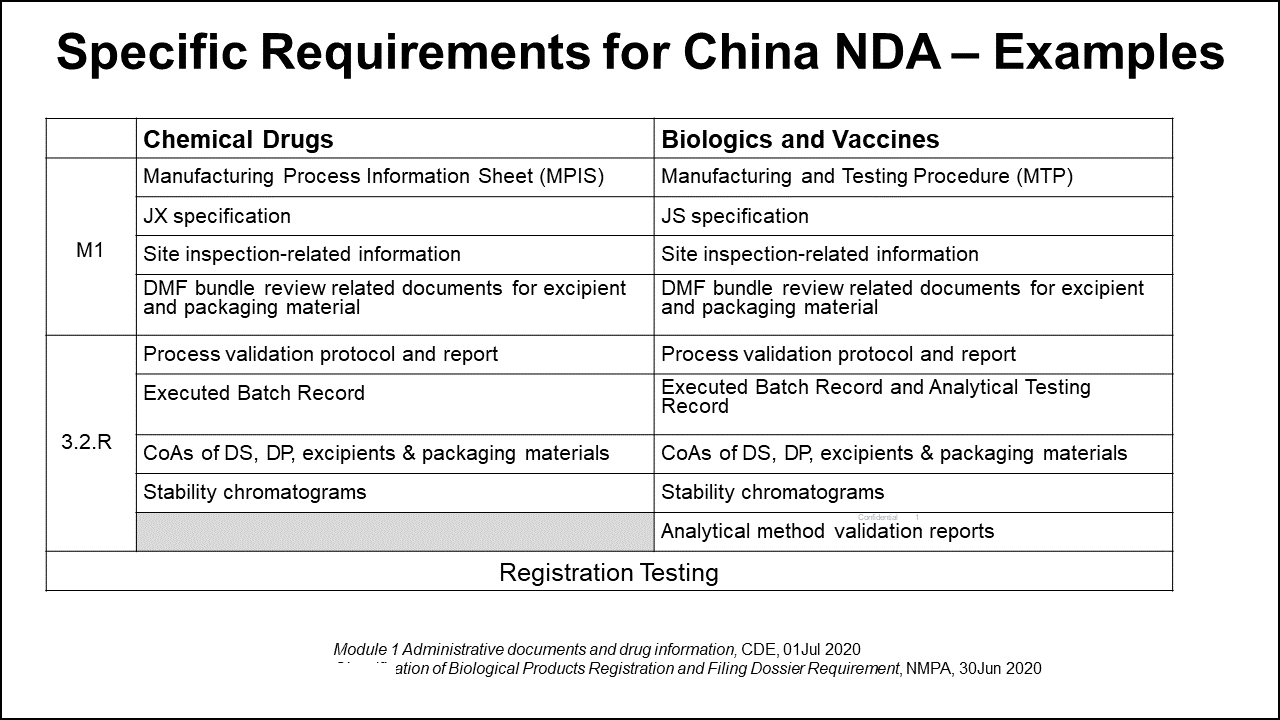
Cao provided some examples of how to navigate CMC submissions, including expectations for specific requirements in the Common Technical Document (CTD) Modules.
Module 1:
- Manufacturing Process Information Sheet (MPIS) and JX specification for chemical drugs—only required in the NDA submission, not required at the CTA stage
- Manufacturing and Testing Procedure (MTP) and JS specification for biological products— required for both CTA & NDA submissions
- Site inspection-related information
- DMF bundle review related documents for excipients and packaging materials
- China Development Safety Update Report (DSUR)
Module 2:
- Complete 2.3.S and 2.3.P required for CTA
Module 3:
- Regional requirements, e.g., stability chromatograms
Cao also provided tables that summarize some of the differences in China, the United States, and the European Union for CTA submissions and New Drug Applications (NDAs) (Figures 5 and 6). Cao characterized some of the China requirements as “challenging,” “potentially prompting IP [intellectual property] concerns,” and “time-consuming” to assemble.

Cao explained that in China the specification is called the JX specification for chemical drugs and the JS specification for biological drugs. “It can be different from the proposed specification in the dossier,” he commented.
For a solid dosage form, China will require a process validation report for the NDA filing, unlike the U.S. FDA.
Registration testing is required. “This is how the JX and JS specifications will be finalized,” Cao explained. “During the registration testing, the company should work with the agency closely to make sure the JX and JS specifications are developed.”
China NDA Approval Letter
Next Cao addressed the China NDA approval letter. NDA approval letters usually have four component parts, which are similar between chemical and biological drugs. Both require label and package insert details and specifications—the JX specification for chemical drugs and the JS specification for biological drugs. For chemical drugs the MPIS is required, and the parallel MTP document must be submitted for biologics.
Note that the attachments are proposed by the applicant at the initial filing, reviewed by the Center for Drug Evaluation (CDE), an agency under the National Medical Products Administration (formerly the China Food and Drug Administration, or CFDA) and approved along with the NDA. They are attached to the NDA approval letter and should be maintained throughout the product lifecycle.
In addition:
- China’s government agencies are involved with review, lab work, and approval by the CDE and NFDC (the testing agency)
- The JX specification for chemical drugs contains controls for drug product
- The JS specification for biologics usually contains controls for the drug substance and drug product
- A combination of release and shelf-life testing items as well as items from the ChP may be added by agency
- The document format should follow the ChP monograph format
- Companies should work closely with government health agencies to ensure the appropriate specifications are established for the China market
- The commitment document requires lifecycle maintenance
The Manufacturing Process Information Sheet (MPIS) document for chemical drugs is required for the NDA but not for the CTA. It contains a detailed summary of the formulation as well as sources and controls of the drug substance, excipients, and packaging materials. It also includes a description of the manufacturing process and the drug product release specification and analytical procedures.
The Manufacturing and Testing Procedures (MTP) document for biologics is required for both the CTA and NDA, although the version in the CTA is less detailed. It also contains a detailed summary of the source and controls of the master and working cell banks, raw materials, excipients, and packaging materials, as well as a detailed description of drug substance and drug product manufacturing processes, specifications, and analytical procedures
…there are multiple layers making up China’s drug regulatory framework…
Note that both documents serve as the basis for onsite inspections and the content must be consistent with the manufacturing process, specifications, and testing procedures at the site. Both are required to be kept current.
“When the company works with agents, trying to finalize the MTP, this could take time, because the discussion and negotiation can be back and forth between the company and the agency,” Cao said.
Cao also provided the scope and documentation requirements for the API, Excipient, and Packaging Material Bundling Review (Figure 7).
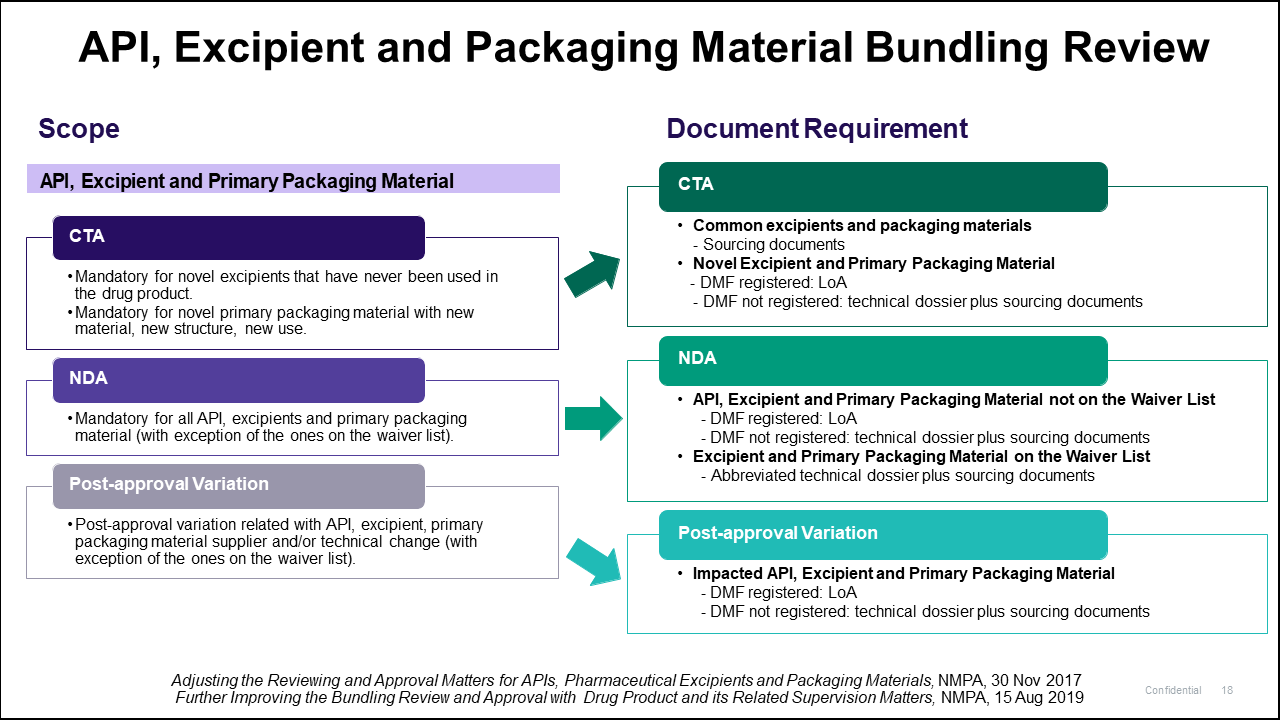
He noted that the Drug Master File (DMF) should be registered on the CDE platform, or if not then a Letter of Authorization (LoA) from the vendor is required. If the DMF is not registered, “it can place multiple challenges on the companies.”
Registration Testing Performed by China Agency Labs
Another important aspect of the drug registration process is laboratory testing. This testing can occur during the NDA, and also can occur during a review of post approval variation. That testing is based on the risk. Before the NDA, it was a standard requirement.
“They offer some selection on when you can apply, but the sample could be a challenge because they need a commercial scale sample. The China specification will be finalized for the NDA part, or it will be updated if it is a post approval variation through this process,” Cao said.
“This is a very important process. The company should work with the China agencies together, including both CDE and NSDC, to make sure the appropriate China specifications will be established,” he emphasized.
Post-Approval CMC Change Management
Cao pointed out that recently multiple post-approval guidelines have come out.
“It is very encouraging because it is getting quite close to the international standards – for example, how major, moderate and final changes are handled (Figure 8).”

Cao pointed out that this guideline on Regulation on Post-marketing Changes of Drugs is quite new (January 2021). “And we still have to practice and see, to accumulate more experience.”
Great Opportunities But Also Challenges
Cao concluded his presentation by summarizing and emphasizing that China has recently:
- Put in place many policies and measures that encourage innovative drug development and global simultaneous development
- Improved review timelines and/or an optimized process for CTA, NDA, and post-approval variations significantly
- Created multiple channels for communication with the health authority to facilitate drug development and improve the dialog with the pharma industry
- Provided for a staged implementation of ICH guidelines to align with international standards
“However,” Cao said, “some specific requirements can be challenging. Thus, driving further global harmonization on regulatory requirements is still particularly important and can further enable China joining global simultaneous development.”
FDA Inspections of API Manufacturing Plants in China Examined
A review of FDA Form 483s issued between 2000-2022 shows that more than 2800 observations were made by FDA investigators during inspections of Active Pharmaceutical Ingredient (API) manufacturing facilities in China.
Redica Systems uses Natural Language Processing (NLP) and Artificial Intelligence (AI) tools called the Quality System Labeling (QSL) model that can query compliance data and map the deficiencies to the quality systems and subtopics within each system. It uses the (6+1) quality systems – the six FDA quality systems plus data integrity.
An analysis of the China API observations using the QSL model reveals the top areas for concern are located within the Production, Laboratory, and Data Integrity quality systems, in that order (see Figure 9, “Distribution of FDA 483 Observations by Quality System).

A deeper analysis shows more specifically the main issues found lacking in subsystems of each of the quality systems.
In the Production System, the most common observations were around Process Validation, Batch Records, and Cleaning Validation subsystems.
Observations for Production System > Process Validation include:
- “process validation…failed to define all significant processing steps”
- “process validation was not performed adequately to support the determination of critical process parameters”
- “equipment qualification is not performed adequately to ensure that equipment operates reproducibly”
- “NMR laboratory instrument used by the contract testing laboratory for the identification test of contaminant in your API has not been qualified”
- “process validation studies and ongoing process monitoring do not include thorough evaluation of variability within a batch or between batches”
Observations for Production System > Batch Records include:
- “individual steps in the batch production records are lumped together preventing the operators and verifiers from documenting completion of the steps as they occur”
- “copies of batch records have no unique sequential identifier”
- “batch records for API drug substance USP lots do not contain samples taken for QC release, retain, and or stability”
- “reported that batch records could not be reviewed before product was released in part because the QA personnel had too large of a workload”
- “we observed your production operator in the process of transcribing and back-dating previously manufactured finished API production batch records”
Observations for Production System > Cleaning Validation include:
- “failure to clean and store equipment and utensils to prevent contamination or carry-over”
- “media used for active air samples settle plates and finger monitoring of personnel do not contain ingredients to neutralize clean room disinfectants”
- “written procedures for cleaning do not have descriptions in sufficient detail of methods and parameters such as volume of water and time”
- “did not conduct risk assessment studies with respect to cleaning validation which address microbiological and or endotoxin contamination”
- “swab sampling performed during your cleaning validation…did not include locations considered to be worst-case”
The primary observations in the Laboratory system were in the areas of Method Validation (no method validation or issues with how it was structured and conducted), handling Out of specification/Out of Trend (OOS/OOT) results (lack of monitoring, lack of documentation or investigation), and the establishment of appropriate laboratory controls (complete records, scientifically sound and appropriate procedures).
Over half of the data Integrity observations pointed to issues with original data – for example:
- “raw data written on a loose-leaf piece of paper was found in an analyst notebook in the QC laboratory”
- “raw data for the assay were not recorded directly into the notebook…not retained”
- “supporting raw data for the validation tests is missing”
- “there is no practice of retaining the original records and as a result there is no information available on what data has been changed or added”
- “maintenance activities are not recorded in equipment use logs as required by your SOP”
In summary, two-thirds of the Form 483 observations evaluated by our Quality Systems Labeling model found in API manufacturing plants in China were found within the Production, Laboratory, and Data Integrity quality systems. It is recommended that anyone doing business with or planning to do business with an API manufacturer in China focus their review on these areas of the company’s manufacturing records and processes as a check on its compliance position.